
维生素E母核合成研究进展*
2015-02-13 09:28柳东明李荀臧恒昌
大学化学 2015年3期
柳东明 李荀 臧恒昌
(山东大学药学院 山东济南250012)
维生素E是一种重要的脂溶性维生素,作为一种生物抗氧化剂,在医药、保健品、食品、化妆品、畜牧业等领域[1]有着非常广泛的应用。维生素E按来源可分为天然维生素E与合成维生素E,市场上约80%的维生素E产品都来自合成[2]。2,3,5-三甲基氢醌(TMHQ)是维生素E的母核结构,与异植物醇经缩合得到维生素E,其生产量是制约维生素E产量的关键因素,而国内目前产能相对不足,工艺上较国外有一定差距。因此,研究适合TMHQ工业化生产的工艺具有较大的应用价值和较广阔的市场前景。
TMHQ的合成工艺在国内外已有多种报道,较早的合成工艺以偏三甲苯为原料[3],先将偏三甲苯氧化为2,3,5-三甲基苯醌(TMBQ),随后还原得TMHQ,但该法因收率偏低无法实现大规模工业生产。另一种早期的合成工艺以均三甲酚为原料[4],在强碱环境下加压氧气氧化均三甲酚为4-羟基-2,4,6-三甲基-2,5-环己二烯酮(TMCH),然后在250℃下进行TMCH甲基转位,还原得TMHQ,收率可达47%(以原料均三甲酚记)。虽然均三甲酚法工艺简单,但是由于原料价格较贵,限制了该工艺的进一步应用。目前,偏三甲苯法与均三甲酚法因收率、成本等问题已逐渐被淘汰,取而代之的是较为先进的TMP法与异佛尔酮法。
1 TMP氧化还原法
TMP法以TMP(2,3,6-三甲基苯酚)为原料,经过氧化得到TMBQ,再经还原制得TMHQ,合成路线如图1。此法由于工艺过程相对简单,原料来源较丰富,TMP的转化率和TMHQ的收率均较高,便于规模化生产,因而得到了广泛的研究[5]。

图1 TMP氧化还原法合成路线
TMP法的关键步骤是TMP的氧化。由于氧化剂、溶剂和催化剂对氧化反应的速率、转化率以及产品收率都有较大影响,因此,如何提高氧化反应的收率成为目前研究的热点。
1.1 氧化反应研究进展
在实验室研究和实际生产中,多以过氧化物或氧气为氧化剂,各种不同方法的区别在催化剂的应用上,根据催化剂的有无与类型可分为无催化剂氧化法、均相催化氧化法以及非均相催化氧化法。
1.1.1 无催化剂氧化法
钱东[6]等利用新的氧化剂体系——醋酸/过氧化氢/盐酸,通过直接氧化法合成得到纯度大于98%的TMHQ。在氧化反应中,以石油醚为溶剂,反应物料醋酸、过氧化氢、盐酸与TMP的物质的量比为6.5:6.5:2.5:1,在回流状态下反应1~1.5h,TMHQ的收率为54.6%。亦可以MnO2-H2SO4混合物为氧化剂[7],通过直接氧化法合成得到TMHQ。比较理想的工艺条件是:H2SO4的含量控制在30%~40%,n(TMP):n(二氧化锰)=1:2,在约65℃反应3h,TMHQ的收率为60.8%。
该类方法使用具有腐蚀性的强质子酸,两步总收率较为可观,但强酸用量较大会造成反应体系体积过大、单一批次产能较低且严重腐蚀设备,故不能成为较完善的工业生产方案。
1.1.2 均相催化氧化法
近几年来,相继报道出多种新型催化剂,旨在节省试剂,避免设备的腐蚀损坏。均相催化中催化剂与原料同处于均一物相,活性中心比较均一,选择性较高,反应动力学一般不复杂。
均相催化剂一般是简单的无机盐,需要在助催化剂存在下才能发挥催化作用。Mçller K等使用FeCl3·6H2O为主催化剂[8],吡啶-2,6-二羧酸和苄胺为助催化剂,以双氧水氧化TMP,转化率大于99%,TMBQ收率79%。
Sun H J[9]等使用催化量的CuCl2,咪唑鎓盐类离子液体为助催化剂,氧气为氧化剂,收率达到86%,并提出了较为全面的氧化机理(图2)。
首先,酚取代氯原子,经过自旋离域形成酚盐自由基;三线态分子氧进攻酚羟基对位形成过氧自由基;随后一价铜化合物进攻过氧基团,通过质子介导的消除,铜簇化合物可以不断再生成为电子受体,同时生成产物TMBQ。
王宪沛[10]等研究发现催化剂MnCl2的用量为TMP物质的量的10%,助催化剂离子液体1-丁基-3-甲基咪唑四氟硼酸盐用量为TMP物质的量的20%,直接通入空气,以其中的氧气为氧化剂,在80℃反应4h,转化率100%,选择性92.6%。
1.1.3 非均相催化法
均相催化剂有难以分离、回收和再生的缺点,并且产品易被催化剂污染。将具有催化活性的金属类化合物通过共价键负载到无机或有机载体上(例如SAB-15[11]、MNL-101[12]),所得非均相催化剂既具有均相催化剂活性高、选择性好的优点,又具有易于分离、能够循环使用的优点优势,部分载体例如干凝胶[13]甚至可以使催化剂在无溶剂情况下发挥作用。

图2 CuCl2催化机理
Trukhan N N等使用含金属钛的介孔中间相硅酸盐催化剂[14],金属钛的含量在1.5%~2%(质量分数),TMBQ选择性86%,TMP转化率100%,显示出钛催化的高活性。
Trubitsyna T A等使用金属钛单取代的杂多酸催化氧化TMP[15],发现TMP与催化剂的物质的量比对控制产物组成至关重要。TMP与催化剂物质的量比为2:1时,联苯酚(BP)收率达到90%,而当TMP与催化剂物质的量比为1:2时,TMBQ收率达到90%。文章猜测该条件下TMP氧化以自由基反应进行,自由基链引发首先要生成酚氧自由基,酚氧自由基在催化位点相对不足时,多发生二聚形成BP,在催化位点相对足够时,酚氧自由基继续自由基链式反应,二聚化副反应有所降低。
为了解TMP氧化的影响因素,从而有针对性地设计高选择性催化剂进行催化氧化,避免副产物的生成,研究人员对TMP的氧化机理做了深入探讨。金属类非均相催化剂应用于苯酚到苯醌的氧化通常被认为有两种机理,一种是涉及双电子路径的异裂机理,另一种是涉及酚氧自由基形成的均裂机理。当以亲水介孔钛硅材料或钛取代的杂多酸盐为催化剂[16-17],双氧水为氧化剂时,氧化反应以均裂机理进行;当使用TS-1或TS-2微孔钛硅催化剂时[18],反应以异裂方式进行。
Kholdeeva O A等合成了多种钛硅催化剂[19],分别将二氯二茂钛、酒石酸二乙基钛、过氧钛复合物连接在二氧化硅材料表面,制得具有单一钛核、双核以及四核的非均相催化剂,并对金属钛介导的催化原理做了深入的研究。对于单核催化剂而言,钛活性中心表面含钛量对产物的选择性有很大影响。钛活性中心表面含钛量范围在0.6~1.0/nm2,TMP主要转化为TMBQ,若含钛量小于0.6/nm2,产物则是TMBQ以及TMP二聚体。双核以及四核催化剂与单核催化剂情况不同,活性中心含钛量的减少对产物的选择性并没有影响。本文从反应机理上解释了双核钛催化二聚副反应较少的原因,见图3。
首先,双钛核活性位点吸附两分子过氧化氢,形成两个位置极为接近的过氧钛基团。一个TMP分子被两个毗邻的过氧钛基团吸附,与第一个过氧钛基团作用形成酚氧自由基,然后被第二个过氧钛基团迅速氧化为TMBQ,因此酚氧自由基的C—C与C—O偶联副产物大大降低,这也是双核钛催化剂性能优于单核钛催化剂的原因。
另一种与金属钛催化不同的是酞菁铁的异裂机理催化。Zalomaeva O V等以酞菁铁作为催化剂[20],过氧叔丁醇为氧化剂,三甲基苯醌收率达到80%。通过GC-MS及1H NMR方法确定了此催化条件下氧化反应的各类副产物。相对于钛硅分子筛—H2O2体系,C—C偶联以及C—O偶联副产物大大减少。本文根据实验结果提出了异裂机理假说,见图4。

图3 钛催化机理
第一步是酚与双铁酞菁配位,酚盐复合物A结合BuOO—形成中间体B,B可以进行O—O键的均裂或异裂。然而,加入自由基捕获剂的EPR实验未显示自由基的明显信号,并且反应动力学实验表明加入自由基捕获剂对TMP的消耗和TMBQ的生成都没有显著影响,因此中间体B的过氧键发生均裂的可能性不大。B的O—O键异裂生成C,由于两个铁原子间电子的离域,C主要以D形式存在。中间体D分子内发生第一次单电子转移形成自由基中间体E,随后发生第二次电子转移,形成阳离子中间体F,BuOO—进攻正电中心构成中间体G。Fe—O配位键断裂,苯环结构脱离酞菁铁形成H,在反应条件下H并不稳定,会很快生成TMBQ。由于异裂反应机理中没有形成酚氧自由基,也就避免了酚氧自由基的C—O与C—C偶联副反应的发生。
非均相催化剂固然有诸多优势,但其存在的较大缺点是水解稳定性差,并且活性会在首次催化后急剧下降。对此,研究人员做了大量工作来解决稳定性问题[21],一些稳定性较好的材料(例如TS-1,Ti-MMM-1、Ti-MMM-2等)相继被报道出来。Ti-MMM-2催化剂结合了高活性与高稳定性,但是对TMBQ的选择性未超过80%,因此还未形成商业竞争力。
在TMP氧化反应中使用催化剂,要综合转化率、选择性、催化剂稳定性与循环利用次数、成本等多种因素,虽然迄今还未有一种催化剂可以兼顾所有因素,但可以肯定的是,TMP的非均相催化氧化具有非常广阔的商业前景。
1.2 还原反应研究进展
相对而言,TMBQ的还原反应较容易实现,其生产方法主要有两类,即化学还原法和催化加氢还原法。
化学还原法即保险粉还原法,朱志庆等[22]使用保险粉水溶液作为还原剂,于40~60℃保温反应,并添加抗坏血酸抗氧化剂,防止TMHQ在后处理过程中氧化,最终TMHQ纯度大于99%,收率95%以上。使用保险粉的优点是后处理简便、收率高;但保险粉的消耗量较大,成本较高。
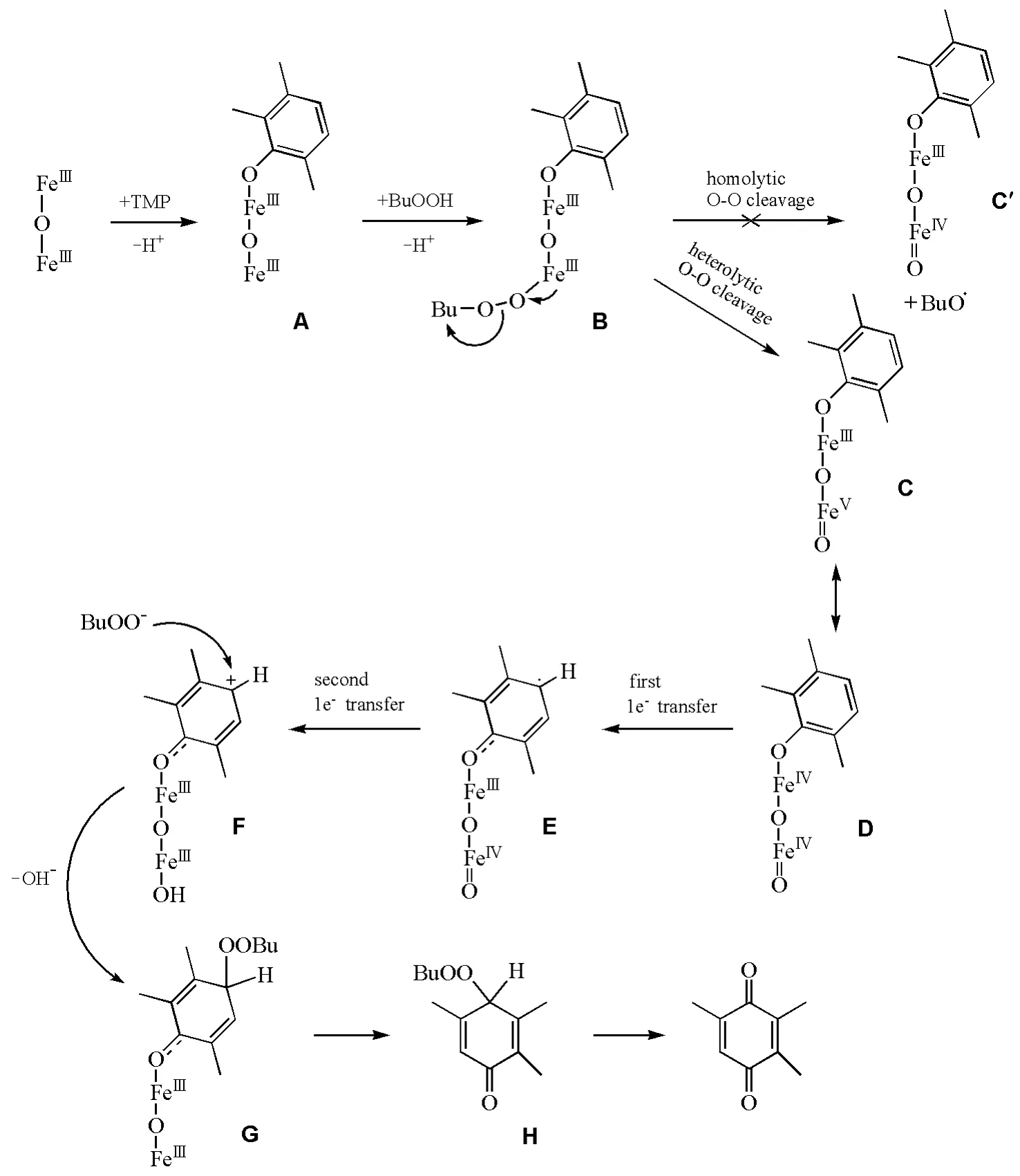
图4 酞菁铁催化机理
催化加氢还原法使用价格便宜的氢气,辅以少量催化剂,实现TMBQ高效还原。钯碳作为常用的高效加氢还原剂,在还原TMBQ中也能发挥很好的效果。张发香等[23]采用5%钯碳为催化剂,乙酸乙酯为溶剂,控制反应压力0.2~1.0MPa进行氢化还原。最终TMHQ含量达到99.4%。
雷尼镍以及贵金属钯、铂是比较常用的高效催化剂,已有文献报道使用这类催化剂可以催化还原TMBQ,得到高收率的TMHQ。
Mukhopadhyay S等使用雷尼镍作为催化剂[24],氢气压力3.5MPa,温度110℃,转化率与选择性均能达到100%。
Tomoyuki Y等使用硅铝负载金属铂[25],40℃常压氢气环境下反应,转化率100%,选择性99.9%,TMHQ纯度大于99%。首次使用催化剂,26min即可完成反应;当催化剂循环次数增多,催化剂活性下降,在循环至第120次时,需要延长反应时间至116min。
杨新礼等[26]采用Pt-Re/Al2O3和Pt-Pd/Al2O3两种双金属催化剂,通过固定床连续工艺,由TMBQ还原得到高收率的TMHQ,并与Pt/Al2O3和Pd/Al2O3催化剂进行了性能比较。结果表明,Pt-Pd/Al2O3催化剂具有较高的初选择性;同时,随着TMBQ空速的提高,Pt-Pd/Al2O3初活性的下降幅度小于Pt/Al2O3和Pd/Al2O3初活性的下降幅度。
2 TMP羟基化法
TMHQ传统的合成方法普遍采用的是氧化还原路线,即先氧化后还原的步骤,实践证明氧化反应较难有效控制,副产物较多,难以达到很高的收率,制约了总收率的提高。近几年来有研究人员打破传统,抛弃氧化还原的思路,应用其他的化学反应替代氧化还原,甚至实现一步成TMHQ。这些新方法虽未实现工业化生产,但其独特的思路为TMHQ生产开辟了一条新道路。
2.1 达金/达夫反应法
不同于传统的先氧化再还原的方法,Kadam S H等使用了全新的路线方法[27],通过达夫反应合成4-羟基-2,3,5-三甲基苯甲醛,再经达金反应得到TMHQ(图5)。达夫反应步骤收率最高可达80%,但需要消耗大量六亚甲基四胺以及需要较高温度;达金反应步骤最高收率可达85%。
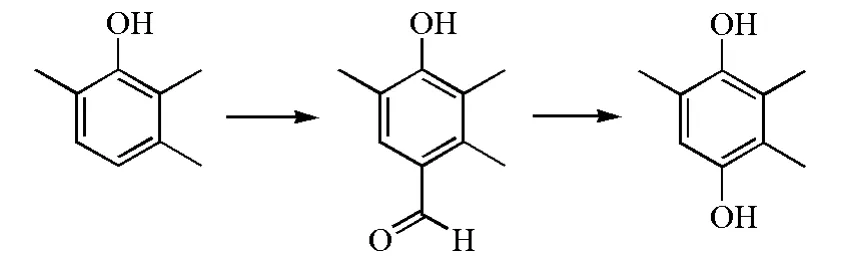
图5 达夫/达金反应路线
2.2 TMP直接羟基化
通过在TMP对位直接羟基化合成TMHQ是非常大胆而且非常具有挑战性的工作。但在芳香环上引入羟基存在一定难度,实验室中传统方案是先引入氨基,再做成重氮盐,酸性条件下水解引入羟基,但这种方案步骤繁琐,收率低,难以实现工业化生产。近几年来已相继报道出不少TMP直接羟基化的研究,这些方法大多需要使用催化剂。
Meng X等使用Cu2(OH)PO4作为羟基化催化剂[28],双氧水作为氧化剂,反应主产物是TMBQ与TMHQ,且对TMHQ的选择性高于TMBQ。当反应在空气中进行,TMHQ选择性达到72.2%;当反应在氮气保护下进行,削弱了TMBQ的氧化,TMHQ选择性可提高到94.7%。
朱虹等[29]采用共沉淀法与柠檬酸法合成了铁酸镁,并用X射线衍射和红外光谱等对不同样品进行了表征。以三甲基苯酚为原料,铁酸镁为催化剂,双氧水为氧化剂,实现了一步羟化为三甲基对苯二酚。文章详细考察了催化剂、温度、溶剂和双氧水等因素对该反应体系的影响。结果表明,铁酸镁具有较好的羟基化催化效果,且催化性能强于常用的羟化催化剂钛硅分子筛等。
Matveeva O以二氧化硅、三氧化二铝、polymers MN-100、Sepabeads EC-HA等材料作为载体[30],将辣根过氧化物酶固定化,使得TMP转化率高达95%,选择性达到98%。此研究首次将酶催化应用到TMHQ的制备中,且取得了很好的效果。酶催化相对于金属络合物催化不仅选择性高,而且不会造成污染,但是生物酶高昂的价格与苛刻的使用环境限制了酶催化的工业化进程。
3 异佛尔酮法
异佛尔酮法首先由丙酮聚合为α-异佛尔酮(α-IP),α-异佛尔酮可异构化为β-异佛尔酮(β-IP),两种异构体均可氧化为茶香酮(KIP),茶香酮重排酰化后得到二乙酸三甲基氢醌酯(DA-TMHQ),随后水解得TMHQ(图6)。由丙酮缩合制备α-异佛尔酮大多在使用催化剂以及一定压力温度条件下进行,其工艺条件相对成熟;工艺路线中较为关键的步骤是异佛尔酮的氧化以及茶香酮的重排酰化。
3.1 异佛尔酮氧化
异佛尔酮的氧化路线分为间接氧化与直接氧化两种,第一条间接氧化路线首先由α-异佛尔酮异构化为β-异佛尔酮,然后氧化β-异佛尔酮制得KIP;第二条路线为直接氧化α-异佛尔酮制备KIP。

图6 异佛尔酮法合成路线
α-异佛尔酮的分子结构中存在共轭效应,导致其稳定性较好,反应活性较低,并使α-异佛尔酮异构化为β-异佛尔酮的过程需要很高的温度,且转化率低;此外,α-异佛尔酮的传统催化氧化基本采用含金属催化反应体系,易对环境造成污染。由于以上因素限制了间接氧化法的推广应用,因此直接氧化α-异佛尔酮制备KIP具有更大的发展前景。
Kishore[31]等应用共沉淀技术合成出铝碳酸镁担载铜、钴、铁固相催化剂,通过X射线粉末衍射及热分析确证了铝碳酸镁的分层网络结构,实验表明铝碳酸镁担载金属铁催化活性最好,α-异佛尔酮转化率达到74%,对KIP的选择性能达到100%。
BAO J Q[32]等首次使用I2作为催化剂,光照5h,条件优化后的结果显示当I2物质的量分数为15%时,α-异佛尔酮转化率达到70.2%,茶香酮选择性达到82.6%。
Wang C M[33]等使用N-羟基邻苯二甲酰亚胺(NHPI)在氧气环境下催化α-异佛尔酮氧化为茶香酮,避开了α-异佛尔酮异构化为β-异佛尔酮的步骤。该氧化反应在无共催化剂存在的条件下于60℃反应10h即可。与传统高温氧化α-异佛尔酮的方法不同,NHPI易回收利用,且催化活性损失很小。该方法转化率较低,只有39%;当使用CuCl2辅助催化[34],可将氧化反应的转化率提高至91.3%,选择性提高到81%。
金属催化剂涉及一系列催化剂的制备与表征,而NHPI结构简单可以直接购得,价格便宜,且不存在重金属污染的问题,相比金属催化剂具有很大优势。
3.2 茶香酮重排酰化
常用的方法是茶香酮与乙酸酐在质子酸或路易斯酸催化下完成,所采用的酸从最初的硫酸、盐酸等,再到后来的氯磺酸、三氟甲基磺酸,收率虽逐步提高,但这些方法需要消耗大量酸,对设备腐蚀严重,甚至还会产生二氧化硫等有毒气体。
维尔纳·邦拉蒂[35]采用三价铟盐(例如三氯化铟、三氟甲烷磺酸铟)作为催化剂,虽然避免了酸腐蚀,异佛尔酮转化率提高到100%,但铟盐高昂的价格与易造成重金属污染限制了其应用。
李浩然等使用酸性离子液体为催化剂[36],其特点是在水中具有较好溶解度,从而可以用水洗的方式将其从反应体系中分离。由于此类酸性离子液体几乎没有蒸气压,热稳定性好,因此在蒸馏催化剂水溶液时这些催化剂毫无损失,最终KIP转化率达到65.8%,DA-TMHQ选择性达到89.5%。
α-异佛尔酮的共轭结构导致分子稳定性较高,曾庆宇[37]等通过改变α-异佛尔酮的分子结构以提高其反应活性,即以α-异佛尔酮为原料,与醋酸酐以及杂多酸类催化剂和空气反应生成酮基异佛尔酮的单酯化产物,在强酸性催化剂及酰化剂存在下反应得到DA-TMHQ,收率72.8%。
4 结论与展望
近年来,维生素E的市场需求量持续增加,使维生素E及其相关化工产品的生产面临前所未有的机遇与挑战。目前国内外已经普遍采用TMP法与异佛尔酮法制备TMHQ,工艺较先进。
虽然工艺路线趋于成熟,但继续研究缩短反应步骤仍然是工艺改进的重点。例如TMP的直接羟基化,代替原来的氧化与还原反应,以及异佛尔酮直接氧化,避开由α-异佛尔酮到β-异佛尔酮的异构化,能大大降低原料与设备成本,简化操作工序。以上两种方式在反应机理层面已被证明是可行的,目前还处于实验室研究阶段,离工业化生产还有一段距离。
另外,科研工作者对原有工艺改进从未间断过。人们对原有TMHQ合成工艺的改进重点集中在各类催化剂的应用上,尤其是在TMP氧化为TMBQ以及异佛尔酮氧化为KIP的反应中应用催化剂。由于氧化反应条件不易控制,反应过程中副产物较多,相应收率降低,应用催化剂的高选择性意在最大限度降低副产物的生成,提高收率。同时催化剂可回收重复利用,在成本控制方面有很大优势。目前已投入应用的催化剂多以金属类催化剂为主,在延长催化剂寿命、降低重金属污染等方面还有待提高,因此发展节能高效的环境友好型催化剂是一项充满挑战又蕴含巨大市场价值的工作。
[1]刘新良.维生素E的连续规模化生产//第三届全国制药工程科技与教育研讨会论文集.杭州:中国药学会,2004,476
[2]孙月婷.广州化工,2011,39(6):34
[3]陈红,吴缨,徐国梅.精细石油化工进展,2002,3(4):25
[4]陈家蓥,王红喜.中国医药工业杂志,1983(5):5
[5]李为民.精细石油化工进展,2005,6(1):38
[6]钱东,王开毅,庞明红.精细化工,1998,15(2):13
[7]钱东,何厚群,王开毅.化学试剂,2002,24(4):231
[8]Mçller K,Wienhçfer G,Schrçder K,et al.Chem Eur J,2010,16(39):10300
[9]Sun H J,Harms K,Sundermeyer J.J Am Chem Soc,2004,126(31):9550
[10]王宪沛,杨瑞云,李文,等.工业催化,2013,21(8):73
[11]孔黎明,周涛,菅盘铭.CN201110106682.0,2011-12-28
[12]Zalomaeva O V,Kovalenko K A,Chesalov Y A.Dalton Trans,2011,40(7):1441
[13]Shishmakov A B,Mikushina Y V,Koryakova O V.Russ J Appl Chem,2011,84(9):1555
[14]Trukhan N N,Romannikov V N,Paukshtis E A,et al.J Catal,2001,202(1):110
[15]Trubitsyna T A,Kholdeeva O A.Kinet Catal,2008,48(3):371
[16]Zalomaeva O V,Trukhan N N,Ivanchikova I D,et al.J Mol Catal A:Chem,2007,277(1-2):185
[17]Kholdeeva O A,Trubitsina T A,Maksimovskaya R I,et al.Ⅰnorg Chem,2004,43(7):2284
[18]Wilkenhöner U,Langhendries G,van Laar F,et al.J Catal,2001,203(1):201
[19]Kholdeeva O A,Ivanchikova I D.Adv Synth Catal,2009,351(11):1877
[20]Zalomaeva O V,Ivanchikova I D,Kholdeeva O A.New J Chem,2009,33(5):1031
[21]Kholdeeva O A,Ivanchikova I D,Guidotti M.Catal Today,2009,141(3):330
[22]朱志庆,袁忠飞,许庆丰.CN201110103349.4,2011-11-16
[23]张发香,张颖,刘明舜.CN201110196916.5,2011-12-28
[24]Mukhopadhyay S.Ore Process Res Dev,2000,4(4):254
[25]Tomoyuki Y.EP87115070.2,1988-4-27
[26]杨礼义,钱东,张茂昆.石化技术与应用,2000,18(2):68
[27]Kadam S,Paknikar S.WO2011/128018 A1,2011-10-20
[28]Meng X J,Sun Z H,Lin S,et al.Appl Catal A-Gen,2002,236(1):17
[29]朱虹,陈诵英.催化学报,2003,24(8):635
[30]Matveeva O,Lakina N,Matveeva V,et al.Top Catal,2011,54(16):1309
[31]Kishore D,Rodrigues A E.Appl Catal A-Gen,2008,345(1):104
[32]Wang C M,Wang G L,Mao J Y.Catal Commun,2010,11(8):758
[33]Chen L H,Tang R R,Li Z Y.Bull Korean Chem Soc,2012,33(2):459
[34]Bao J Q,Chen L H,Tang R R.J Cent South Univ,2012,19(10):2755
[35]维尔纳·邦拉蒂.CN200480035561.X,2006-12-27
[36]李浩然,陈志荣,胡兴邦,等.CN200810062439.1,2009-12-23
[37]曾庆宇,宋文杰,张琴.CN201110061894.1,2011-9-14
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
能源化工(2021年2期)2021-12-30
第一财经(2019年8期)2019-08-26
中国调味品(2017年2期)2017-03-20
广东石油化工学院学报(2016年6期)2016-05-17
哈尔滨医药(2015年2期)2015-12-01
学习月刊(2015年14期)2015-07-09
中学化学(2015年2期)2015-06-05
合成化学(2015年8期)2015-04-23
合成化学(2015年5期)2015-03-26
