
Genetic Analysis of the VP2 Hypervariable Region of Thirty-six Infectious Bursal Disease Virus Isolates in China during 2009-2012
2015-02-06 08:44XiaoleQILitingQINYulongGAOHongleiGAOYingyingLILiGAOZhenLUNianWANGYumingCHENLizhouZHANGKaiLIYongqiangWANGXiaomeiWANG
Xiaole QI,Liting QIN,Yulong GAO,Honglei GAO,Yingying LI,Li GAO,Zhen LU,Nian WANG,Yuming CHEN,Lizhou ZHANG,Kai LI,Yongqiang WANG,Xiaomei WANG,2**
1.Division of Avian Infectious Diseases,State Key Laboratory of Veterinary Biotechnology,Harbin Veterinary Research Institute, the Chinese Academy of Agricultural Sciences,Harbin 150001,China;
2.Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Disease and Zoonoses,Yangzhou 225009,China;
3.Shandong New Hope Liuhe Co.,Ltd,Qingdao 266000,China
Genetic Analysis of the VP2 Hypervariable Region of Thirty-six Infectious Bursal Disease Virus Isolates in China during 2009-2012
Xiaole QI1*,Liting QIN1,3*,Yulong GAO1,Honglei GAO1,Yingying LI1,Li GAO1,Zhen LU1,Nian WANG1,Yuming CHEN1,Lizhou ZHANG1,Kai LI1,Yongqiang WANG1,Xiaomei WANG1,2**
1.Division of Avian Infectious Diseases,State Key Laboratory of Veterinary Biotechnology,Harbin Veterinary Research Institute, the Chinese Academy of Agricultural Sciences,Harbin 150001,China;
2.Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Disease and Zoonoses,Yangzhou 225009,China;
3.Shandong New Hope Liuhe Co.,Ltd,Qingdao 266000,China
[Objective]Infectious bursal disease(IBD)is a highly contagious immunosuppressive disease caused by infectious bursal disease virus (IBDV).IBDV is genetically prone to mutation,which results in challenges to the disease prevention and control.Thus,it is necessary to continuously monitor the prevalence of IBDV. [Method]36 IBDVs were identified from ten provinces in China from 2009 to 2012. Partial fragments of VP2,including the hypervariable region (HVR),from new isolates were sequenced and analyzed through comparisons with published sequences of IBDV,including 18 strains isolated previously by our lab and 24 reference strains from China and around the world.[Result]Phylogenetic analysis showed a co-existence of IBDV strains belonging to classic,variant,attenuated,and very virulent IBDV(vvIBDV)in China.vvIBDVs remain the predominant strains in China and the new subgroup was emerging.Alignment analysis revealed several distinct amino acid mutations that might be involved in virulence or antigenicity variation.[Conclusion]The results offered evolutionary clues showing the emerging trend of obvious variations and diversity of IBDV in major poultry-producing regions of China particularly in recent years.These findings will contribute to a better understanding of the genetic evolution of IBDV.
Genetic analysis;VP2;Infectious bursal disease virus
I nfectious bursal disease virus(IBDV)is the causative agent of a highly contagious immunosuppressive disease in chickens.Infectious bursal disease (IBD)poses a great threat to the poultry industry worldwide[1].There are two serotypes of IBDV:serotype I (pathogenic to chickens)and II(nonpathogenic).Similar to other RNA viruses,IBDV has a high mutation rate during viral replication.During the past 50 years,the classic,antigenic variant,and very virulent IBDV (vvIBDV)strains have emerged in succession,presenting new challenges for the effective prevention and control of IBD[2].
As a member of Avibirnavirus of the Birnaviridae family,IBDV contains a double-stranded RNA genome including two segments(A and B)within a non-enveloped icosahedral capsid[1]. Segment A encodes a nonstructural viral protein 5(VP5)and a polyprotein precursor NH2-pVP2-VP4-VP3-COOH that can be self-cleaved into VP2,VP4,and VP3[3].Segment B encodes VP1(the viral RNA-dependentRNA polymerase),which is responsible for viral genome replication and RNA synthesis[4].VP2 is the only capsid protein of the icosahedral capsid[5-6], which is responsible for virulence,cell tropism[7-12],antigenic variation,and contains neutralizing antibodies-inducing epitopes of IBDV[2,13-14].The length of VP2 is 441 amino acids (aa).The VP2 region of 206-350 aa is identified as the hypervariable region(HVR)and contains four hydrophilic regions:210-225(peak A),247-254(minor peak 1), 281-292(minor peak 2),and 312-324 (peak B)[7,14].VP2 is folded into three distinct domains,designated base(B), shell(S),and projection (P)[3,15-16]. The tower-like variable P domain contains four loops:PBC(204-236 aa of VP2),PDE(240-265 aa),PFG(270-293 aa),and PHI(305-337 aa)[5].VP2 is under more selective pressure than the other regions of the IBDV genome [1 3].Thus,VP2 has the highest mutation frequency and has been most often used for the molecular characterization of IBDV[17-22].
Since the first report of IBD in china in 1982[23],IBDVs have spread widely throughoutmany provinces, causing economic losses to the poultry industry[22,24-29].Although vaccination contributes mostly to the prevention and control of IBD,new outbreaks of IBD have continued to occur.It is necessary to continuously monitor the prevalence of IBDV.To monitor the newly emerging strains and to assess the extent of genetic diversity in China, focusing on the hypervariable region of VP2,36 IBDV strains collected from the major regions of poultry production were sequenced and analyzed.
Materials and Methods
Clinical samples
Thirty-six atrophic and hemorrhagic bursa tissues were collected from different commercial broiler and layer farms exhibiting the clinical and pathologic features ofIBD in ten provinces of China during 2009-2012 (Table 1).The bursa tissues were used to generate a 10% (wt/vol)homogenate in phosphate buffered saline (pH 7.2)with penicillin and streptomycin.The homogenate was frozen and thawed three times,and the supernatant was harvested by centrifugation at 5 000 x g and 4℃for 5 min.
Viral RNA extraction and RT-PCR
Viral RNA extraction from the bursa homogenates was performed using the PurelinkTMRNA Mini kit(Invitrogen,Carlsbad,CA,USA)following the instructions of the manufacturers. Viral cDNA was synthesized using the M-MLV Reverse Transcriptase kit(Invitrogen,Carlsbad,CA,USA)according to the manufacturer’s instructions. With reference to the sequences of the vvIBDV Gx strain(GenBank accession number DQ403248),a pair of primers, A628U (5’-CCTCAGCTTACCCACATC-3’)and A1540L(5’-CCTTCCCCAATTGCATGG-3’), were designed using the OLIGO 6.0 software. A VP2 fragment(628-1 557 bp of segment A) was amplified with PrimeSTARTM HS DNA Polymerase (Takara Biotechnology Co., Ltd., Dalian,China)using the following program:initial denaturation at 95℃for 5 min;35 cycles consisting of denaturation for 30 s at 94℃,annealing for 30 s at 56℃,and elongation for 1 min at 72℃;and final elongation at 72℃for 10 min.
Sequencing
The RT-PCR products were purified with the BioSpin Gel Extraction Kit (Bioer Technology Co.,Tokyo,Japan) and sequenced by Beijing Liuhe Huada Gene Technology Company(Bei-jing,China).A 777-bp VP2 fragment sequence(547-1 323 bp)corresponding to 183-441 aa,including the hypervariable region(HVR,206-350 aa), was identified and submitted to Gen-Bank(Table 1).
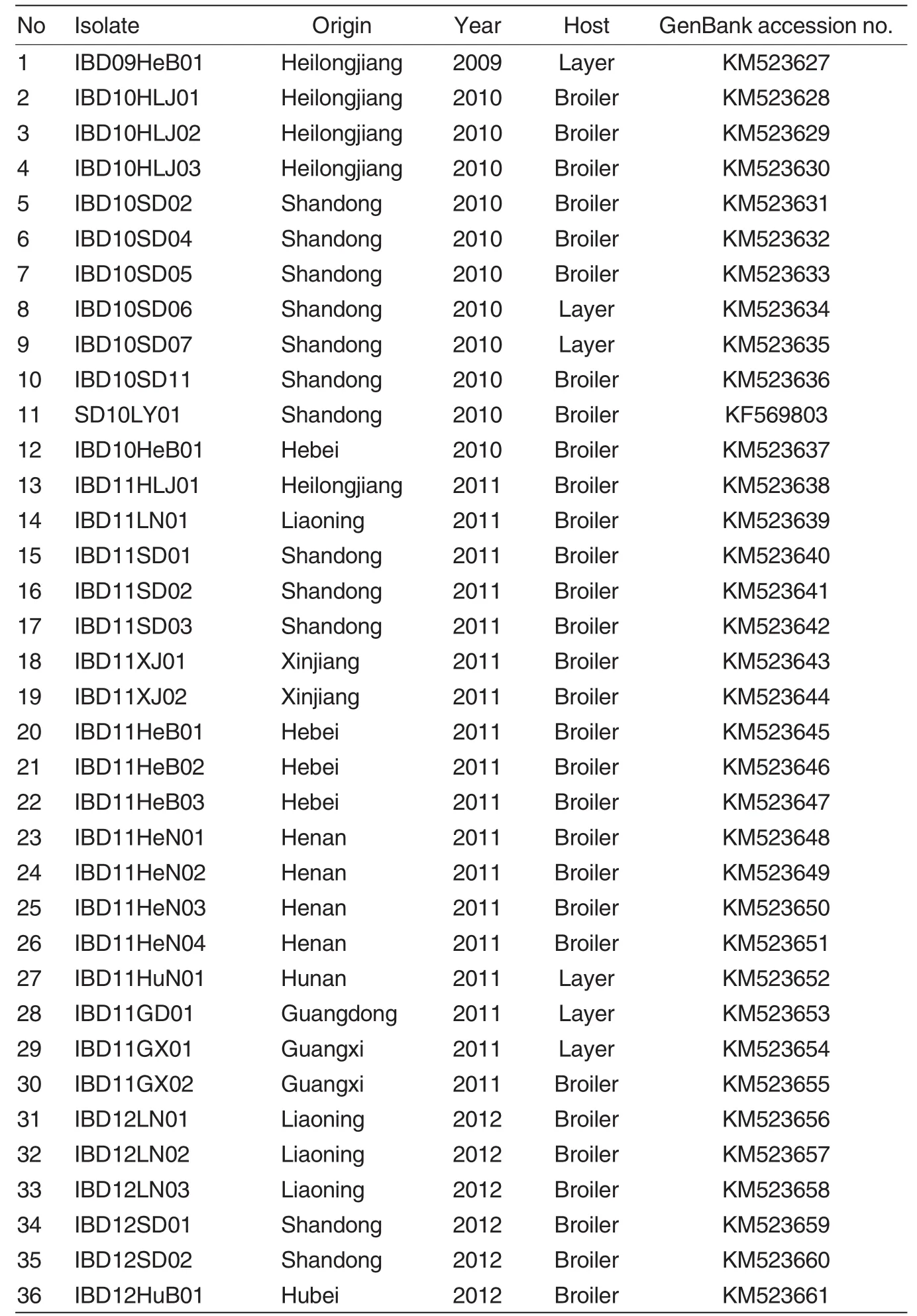
Table 1 IBDV strains isolated in this study
Sequence analysis
A partial fragment of VP2 including the HVR mentioned above was selected for sequence analysis.In addition to the 36 sequences mentioned above (Table 1),18 sequences of IBDV strains isolated previously by our lab and 24 sequences of IBDV reference strains obtained from GenBank were also selected for sequence analysis (Table 2).As for their origins,63 of the Chinese strains were isolated from the following 16 provinces or districts:Heilongjiang,Liaoning,Shandong,Hebei,Henan,Hubei,Hunan, Shanghai,Jiangsu,Fujian,Zhejiang, Guangdong,Guangxi,Yunan,Xinjiang,and Hongkong.The phenotypes and sequence accession numbers of these IBDV strains are summarized in Table 2.Alignment and phylogenetic analyses based on the nucleotide or amino acid sequenceswere performed using the Clustal W program (version 1.8)[30]and the MEGA program (version 3.1)[31],and the confidence levels were assessed using 1 000 bootstrap replications.
Nucleotidesequenceaccession numbers
The sequences obtained in this study have been deposited in Gen-Bank under the accession numbers shown in Table 1.
Results
Division of the Chinese isolates into four branches
The sequences of all 36 isolates from the clinical samples and other 27 Chinese isolates from GenBank examined in this study belonged to serotype I of IBDV,which had an identity of 94.7%-99.5%with the UK661 strain (serotype I),and 76.9%-79.4%identity with the 23/82 strain (serotype II). The Chinese isolates were distinctly divided into four major branches:classic strains,variant strains,very virulent strains,and attenuated strains(Fig.1). Most of the sequences (33/36)from the clinical samples in this study belonged to the very virulent branch. IBD11LN01 and IBDXJ01 formed a distinct branch with variant strains of GLS and Variant E.IBD12SD02 had close homology with the classic strain of IM(98.7%).
Classification of two subgroups of the very virulent branch
In the very virulent branch,33 sequences from the clinical samples investigated in this study were scattered in two subgroups(Fig.2).Most of the Chinese strains formed a distinct subgroup I and were closely related to the reference strains UK661 and D6948 of vvIBDV.The other 13 new isolatesfrom the clinical samples collected during 2010-2012 and four previous isolates were branched out of subgroup I and formed a new subgroup II, which included strains scattered in different regions of China.
Molecular characteristics of IBDV strains
Thededucedaminoacidsequences of the strains isolated in this study were aligned and compared with reference strains including vvIBDV UK661.Eight amino acid residues in VP2 (222A,242I,253Q,256I,279D, 284A,299S,and 330S),which are usually conserved in vvIBDV strains, were also found in almost all Chinese isolates in the vvIBDV branches showed in Fig.2.Interesting mutations were observed in some strains.In subgroup I, IBD11HeN04 and IBD10HLJ03 had two interesting mutations,Q253H and A284T,compared with UK661,and these mutations are key determinants of cell-tropism and virulence[10,12,31-32],and another mutation I294V or S330R was also found in these strains.IBD10HLJ02 had two mutations I272T and D279N.In subgroup II,16 out of 17 strains exhibited the specific mutation D212N,whereas 15 out of 17 strains had the specific mutation V384I.In addition,the mutations A222T and I308L were observed in IBD10SD02 and IBD10HLJ02 of subgroup II,respectively.
Discussion
vvIBDV have been identified as the predominant prevalent viruses in China[22,24-29].To further understand the recent prevalence of IBD,36 IBDV strains isolated from 10 provinces from 2009 to 2012 were analyzed.The results showed that the sequences of 92%of the strains(33/36)from clinical samples were closely related to vvIBDV strains(Fig.1),which indicated that vvIBDV-like strains are still predominantly circulating in the major regions of poultry production in China.
As an RNA virus,IBDV has the potential for rapid evolution.To more fully understand the variability among relatively recent isolates of vvIBDV,54 Chinese strains,including 33 recent isolates,were subjected to phylogenetic analysis based on the nucleotide acid sequence of a partial fragment of VP2 including the HVR.All of the isolates from 16 provinces of China could be divided into two subgroups according to the phylogenetic analysis (Fig.2).In addition,68.5% (37/54)of the Chinese strains clustered together and formed subgroup I,indicating that they are closely related to vvIBDV strains of Asian/European lineage.It is worth noting that many new isolates in China,particularly during 2010-2012, were branched out of subgroup I and formed the new subgroup II.The results offer evolutionary clues showing the vvIBDV strains in China tended to exhibit diversity,particularly in recent years.In most cases,the geographical pattern of IBDV was overlapped likely because of the strength of the economic integrative trend.Subgroup II was a mixed subgroup that included strains from north,central,and south China.The complicated popularity of IBDV may speed up viral evolution and mutation,which would present an increasing challenge to disease control.
The genetic variability resulting from mutations in RNA viruses has long been considered fundamental to their evolution,adaptation,and escape from host responses[33].Because of selective pressure,most exchanges of amino acid residues have occurred in the capsid protein VP2 of IBDV,which could be immediately exposed to the immune system[5,13].Sequence alignment showed that a few mutations have occurred in recent vvIBDV-like isolates and that all of these were focused on the hypervariable region of VP2.Residues 253 and 284,which are located in the hydrophilic loops PDEand PFGat the tip of the VP2 spikes, have been reported to be responsible for cell-tropism and virulence[8-12,32]. Compared with the reference vvIBDV strain UK661, the new strains IBD11HeN04 and IBD10HLJ03 have two interesting mutations Q253H and A284T even though they were clustered into vvIBDV in the phylogenetic tree.It has been verified that the combination of the mutations Q253H and A284T may result in adaptation to cell culture and attenuated vvIBDV[10]. Jackwood and Stoute recently identified some strains in northeast Ohio with a special residue E at position 253 that could break through the maternal immunity[34].In addition,the mutations I294V and S330R were also found in IBD11HeN04 and IBD10HLJ03,respectively.Residue 330 is located at the interface between subunits in the trimeric spike of VP2,and its mutation might lead to subtle alterations in the jelly roll,ultimately leading to a differ-ent conformation of loop PBC[14].It was recently reported that the recognition of the monoclonal antibody 57(MAb 57)epitope involves the PHIloop and a particular residue in position 330[14]. Usually,there are conserved amino acid differences:294I and 330S for virulent strain,and 294V and 330R for attenuated strains.Further identification using animal experiments are required to determine whether IBD11HeN04 and IBD10HLJ03 may be naturally attenuated IBDV.
Residue 222 of VP2 is also one of the interesting mutation sites of IBDV, A222T was observed in the IBD10SD02 strain.In most cases, residue 222 is usually A in vvIBDV and P in non-vvIBDV strains.Occasionally, 222T of VP2 was observed in some variant strains,namely DEL-E,GLS, and GA988[14,35].In the Belg strain,the new occurring mutation P222S was found[14].It has been reported that residue 222 is closely related with viral antigenicity because P222S or A222T could change the reactivity of the responding epitope with MAb 67[14].The characteristics of residues 222,249, 286,and 318 were recently used to perform a phylogenetic analysis of IBDV,which suggested recombination events between variant and classic IBDV[36].Because of the natural mutation T222A,vaccine strain Del-E even broke through the maternal immunity provided by parenteral vaccination[35]. Another mutation,D279N,was found in the IBD10HeB01 strain,which was located on the hydrophilic loop PFGof VP2[5].Lim et al.reported that the double mutations D279N/A284T could change the cell-tropism of vvIBDV[9]. However,Brandt et al.reported that D279N/A284T could not adapt IBDV to CEF[8].To date,the involvement of residue 279 in the virulence of IBDV has not been investigated.In addition, several other mutations that are characteristic of new isolates in this study were observed,such as D212N in the hydrophilic loop PBC,I272T in PFG, I308L in PHI,and V384I in the N terminus of VP2.It has been reported that the mutations at positon 212 are becoming common among new isolates of IBDV in south China[37].Our study showed that these types of mutated IBDV may have been successfully transmitted to north China. Loops PDEand PFGcontribute to celltropism and virulence,whereas PBCand PHI contain the virus-neutralizing epitopes[5].It would be interesting to further investigate the biological significances of these amino acid mutations in detail.
It has been reported that there is a co-existence of epidemiologically mixed IBDV strains with different characteristics in many countries[18,38].In this study,Fig.1 showed a mixed popularity with classic,variant,attenuated, and very virulent IBDV strains in China.In recent years,classic and variant IBDVs have been seldom reported in China.In this study,the IBD12SD02 strain with its own special amino acid 217L/242V/270T from Shandong Province had 98.7%identity with the classic reference strain of IM.Variant strains were also found in Liaoning province (IBD11LN01)and Xinjiang district (IBD11XJ01), respectively. Compared with the reference variant strains GLS and variant E,all five distinctive amino acid exchanges in the IBD11LN01 and IBD11XJ01 strains occurred in the hydrophilic regions of VP2:D213N and Q221K located at the hydrophilic peak A,T286I located at the hydrophilic minor peak 2,and G318D and D323E located at peak B (Fig.3).These regions are under selection pressure because they are immediately exposed to the immune system[5,13].Further experiments are necessary to identify whether these changes result in differences in the antigenicity of IBDV.
In summary,the results show a co-existence of epidemiologically mixed infection including classic,variant,attenuated,and very virulent IBDVs in China.vvIBDV are popular strains and new variations tend to be emerging in the major regions of poultry production,particularly in recent years.These findings will contribute to a better understanding of the genetic variation and diversity of IBDV.
[1]MULLER H,ISLAM MR,RAUE R.Research on infectious bursal disease: The past,the present and the future[J]. Vet Microbiol,2003,97:153-165.
[2]JACKWOOD DJ,SREEDEVI B,LEFEVER LJ,et al.Studies on naturally occurring infectious bursal disease viruses suggest that a single amino acid substitution at position 253 in VP2 increases pathogenicity[J].Virology,2008,377: 110-116.
[3]BIRGHAN C,MUNDT E,GORBALENYA AE.A non-canonical Lon proteinase lacking the ATPase domain employs the Ser-Lys catalytic dyad to exercise broad control over the life cycle of a double-stranded RNA virus[J]. EMBO J,2000,19:114-123.
[4]VON EINEM UI,GORBALENYA AE, SCHIRMEIER H,et al.VP1 of infectious bursal disease virus is an RNA-dependent RNA polymerase[J].J Gen Virol,2004,85:2221-2229.
[5]COULIBALY F,CHEVALIER C,GUTSCHE I,et al.The birnavirus crystal structure reveals structural relationships among icosahedral viruses [J].Cell, 2005,120:761-772.
[6]SAUGAR I,LUQUE D,ONA A,et al. Structural polymorphism of the major capsid protein of a double-stranded RNA virus:An amphipathic alpha helix as a molecular switch [J].Structure, 2005,13:1007-1017.
[7]BOOT HJ,TER HUURME A,HOEKMAN AJW,et al.Rescue of very virulent and mosaic infectious bursal disease virus from cloned cDNA:VP2 is not the sole determinant of the very virulent phenotype[J].J Virol,2000,74: 6701-6711.
[8]BRANDT M,YAO K,LIU MH,et al. Molecular determinants of virulence, cell tropism,and pathogenic phenotype of infectious bursal disease virus[J].J Virol,2001,75:11974-11982.
[9]LIM BL,CAO YC,YU T,et al.Adaptation of very virulent infectious bursal disease virus to chicken embryonic fibroblasts by site-directed mutagenesis of residues 279 and 284 of viral coat protein VP2[J].J Virol,1999,73:2854-2862.
[10]QI X,GAO H,GAO Y,et al.Naturally occurring mutations at residues 253 and 284 in VP2 contribute to the cell tropism and virulence of very virulent infectious bursal disease virus[J].Antivira Res,2009,84:225-233.
[11]QI X,ZHANG L,CHEN Y,et al.Mutations of residues 249 and 256 in VP2 are involved in the replication and virulence of infectious bursal disease virus[J].Plos One,2013,8:e70982.
[12]VAN LOON A,DE HAAS N,ZEYDA I, et al.Alteration of amino acids in VP2 of very virulent infectious bursal disease virus results in tissue culture adaptation and attenuation in chickens [J].J Gen Virol,2002,83:121-129.
[13]DURAIRAJ V,SELLERS HS,LINNEMANN EG,et al.Investigation of the antigenic evolution of field isolates us-ing the reverse genetics system of infectious bursal disease virus(IBDV) [J].Arch Virol,2011,156:1717-1728.
[14]LETZEL T,COULIBALY F,REY FA,et al.Molecular and structural bases for the antigenicity of VP2 of infectious bursal disease virus [J].J Virol,2007, 81:12827-12835.
[15]GARRIGA D,QUEROL-AUDI J,ABAITUA F,et al.The 2.6-angstrom structure of infectious Bursal disease virusderived T=1 particles reveals new stabilizing elements of the virus capsid [J].J Virol,2006,80:6895-6905.
[16]LEE CC,KO TP,CHOU CC,et al. Crystal structure of infectious bursal disease virus VP2 subviral particle at 2.6 angstrom resolution:Implications in virion assembly and immunogenicity [J].J Struc Biol,2006,155:74-86.
[17]ALFONSO-MORALES A,MARTINEZPEREZ O,DOLZ R,et al.Spatiotemporal phylogenetic analysis and molecular characterisation of infectious bursal disease viruses based on the VP2 hyper-variable region[J].Plos One,2013,8:e65999.
[18]ISLAM MT,THANH HOA L,RAHMAN MM,et al.Molecular characterization of two Bangladeshi infectious bursal disease virus isolates using the hypervariable sequence of VP2 as a genetic marker[J].J Vet Sci,2012,13:405-412.
[19]JACKWOOD DJ,SOMMER-WAGNER S.Genetic characteristics of infectious bursal disease viruses from four continents.Virology,2007,365:369-375.
[20]LEVIN BR,LIPSITCH M,BONHOEFFER S.Evolution and disease-Population biology,evolution,and infectious disease:Convergence and synthesis [J].Science,1999,283:806-809.
[21]YAMAGUCHI T,KASANGA CJ,TERA-SAKI K,et al.Nucleotide sequence analysis of VP2 hypervariable domain of infectious bursal disease virus detected in Japan from 1993 to 2004[J]. J Vet Med Sci,2007,69:733-738.
[22]YUWEN Y,GAO Y,GAO H,et al.Sequence analysis of the VP2 hypervariable region of eight very virulent infectious bursal disease virus isolates from the northeast of china[J].Avian Dis, 2008,52:284-290.
[23]ZHOU J,LIU FZ,CHEN LY.Isolating of infectious bursal disease virus from Beijing[J].Chin J Vet Med,1982,7: 25-26.
[24]CAO YC,YEUNG WS,LAW M,et al. Molecular characterization of seven Chinese isolates of infectious bursal disease virus:Classical,very virulent, and variant strains[J].Avian Dis,1998, 42:340-351.
[25]GAO H,WANG X,GAO Y,et al.Direct Evidence of reassortment and mutant spectrum analysis of a very virulent infectious bursal disease virus[J].Avian Dis,2007,51:893-899.
[26]HE X,WEI P,YANG X,et al.Molecular epidemiology of infectious bursal disease viruses isolated from Southern China during the years 2000-2010[J]. Virus Genes,2012,45:246-255.
[27]LIU D,ZHANG X,YAN Z,et al.Molecular characterization and phylogenetic analysis of infectious bursal disease viruses isolated from chicken in South China in 2011[J].Trop Anim Health Prod,2013,45:1107-1112.
[28]Qi X,Gao L,Qin L,et al.Genomic sequencing and characterization of a very virulent strain of infectious bursal disease virus isolated in China[J].Agric Sci Technol,2011,12:1950-1953.
[29]WANG YS,WANG ZC,TANG YD,et al.Comparison of four infectious bursal disease viruses isolated from different bird species[J].Arch Virol,2007, 152:1787-1797.
[30]THOMPSON JD,GIBSON TJ,PLEWNIAK F,et al.The CLUSTAL_X windows interface:flexible strategies for multiple sequence alignment aided by quality analysis tools[J].Nucleic Acids Res,1997,25:4876-4882.
[31]KUMAR S,TAMURA K,NEI M.MEGA3:Integrated software for molecular evolutionary genetics analysis and sequence alignment.BriefBioinform, 2004,5:150-163.
[32]Mundt E.Tissue culture infectivity of different strains of infectious bursal disease virus is determined by distinct amino acids in VP2[J].J Gen Virol, 1999,80:2067-2076.
[33]MOYA A,ELENA SF,BRACHO A,et al.The evolution of RNA viruses:A population genetics view[J].Proc Natl Acad Sci USA,2000,97:6967-6973.
[34]JACKWOOD DJ,STOUTE ST.Molecular evidence for a geographically restricted population of infectious bursal disease viruses[J].Avian Dis,2013, 57:57-64.
[35]JACKWOOD DJ,SOMMER-WAGNER SE.Amino acids contributing to antigenic drift in the infectious bursal disease Birnavirus(IBDV)[J].Virology, 2011,409:33-37.
[36]JACKWOOD DJ.Molecular Epidemiologic Evidence of homologous recombination in infectious bursal disease viruses[J].Avian Dis,2012,56:574-577.
[37]HE XM,XIONG ZX,Yang L,et al.Molecular epidemiology studies on partial sequences of both genome segments reveal that reassortant infectious bursal disease viruses were dominantly prevalent in southern China during 2000-2012[J].Arch Virol.2014,159: 3279-3292.
[38]IGNJATOVIC J,SAPATS S.Confirmation of the existence of two distinct genetic groups of infectious bursal disease virus in Australia[J].Aust Vet J, 2002,80:689-694.
Responsible editor:Xiaoxue WANG
Responsible proofreader:Xiaoyan WU
36株鸡传染性法氏囊病病毒中国分离株 (2009~2012)VP2基因高变区的序列分析
祁小乐1*,秦立廷1,3*,高玉龙1,高宏雷1,李颖颖1,高立1,卢珍1,王念1,陈玉明1,张礼洲1,李凯1,王永强1,王笑梅1,2**
(1.中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室禽传染病研究室,黑龙江哈尔滨150001;2.江苏省动物重要疫病与人畜共患病协同创新中心,江苏扬州 225009;3.山东新希望六合有限公司,山东青岛 266000)
[目的]鸡传染性法氏囊病(IBD)是由传染性法氏囊病病毒(IBDV)引起的一种高度接触性免疫抑制性传染病。IBDV易变异,给防控提出了挑战。持续监测IBDV的流行十分必要。[方法]对36株源于2009~2012年我国10个省的IBDV毒株的VP2部分基因 (包含高变区)进行了扩增、测序。将其与本实验室早期分离的18株IBDV,以及源自我国和世界其它地区的24株参考毒株,进行序列比对分析。[结果]IBDV经典毒株、变异毒株、弱毒株和超强毒株(vvIBDV)在我国同时存在。vvIBDV仍然是我国的主要流行毒株,而且新的亚群正在形成。序列比对分析显示,这些毒株中存在可能与毒力和抗原变异相关的氨基酸突变。[结论]该研究有助于笔者进一步认识IBDV的遗传变异。
遗传分析;VP2;传染性法氏囊病病毒
国家自然科学基金重点项目(31430087);哈尔滨市应用技术研究与开发基金(2014AB3AN058);哈尔滨市科技创新人才基金(2014RFQYJ129);现代农业产业技术体系建设专项基金(nycytx-42-G3-01).
祁小乐(1980-),男,河南宜阳人,博士,副研究员,硕士生导师,研究方向为禽免疫抑制 病,E-mail:qxl@hvri.ac.cn; 秦 立 廷(1981-),男,山东新泰人,博士,副研究员,研究方向为动物疫病防控,E-mail:qinlt2013@163.com。*祁小乐和秦立廷为同等贡献作者。**王笑梅为通讯作者,博士,研究员,博士生导师,研究方向为禽免疫抑制病,E-mail:xmw@hvri.ac.cn。
2015-06-05
修回日期 2015-09-16
Supported by National Natural Science Foundation of China (No.31430087);the Application Technology Research and Development Fund of Harbin (no. 2014AB3AN058);the Special Fund for Scientific and Technological Innovative Talents of Harbin (No.2014RFQYJ129);the Modern Agro-industry Technology Research System of China(No.nycytx-42-G3-01).
*The authors contributed equally to this study.
**Corresponding author. E-mail: xmw@hvri.ac.cn Received: June 5, 2015 Accepted: July 16, 2015
猜你喜欢
吉林畜牧兽医(2022年2期)2022-11-16
广东海洋大学学报(2022年2期)2022-03-31
井冈山大学学报(自然科学版)(2022年2期)2022-03-31
河南科技学院学报(自然科学版)(2022年1期)2022-02-25
中国农业大学学报(2022年2期)2022-01-05
创新作文(1-2年级)(2018年12期)2018-04-24
中国实用医药(2016年4期)2016-02-23
吉林农业·下半月(2014年7期)2014-09-23
课外阅读(2011年24期)2011-12-28
故事作文·低年级(2009年9期)2009-12-10
 Agricultural Science & Technology2015年8期
Agricultural Science & Technology2015年8期
- Agricultural Science & Technology的其它文章
- Establishment of a Method for Determination of Anemoside B4 Content in Pulsatilla Water Extract
- Screening and Evaluation of Different Wheat Cultivars for Resistance to Sitobion avenae at Seedling and Adult-plant Stages
- Interspecific Cross Compatibility of Rhododendron in Changbai Mountain
- Variations of Frost-free Period and Its Impact on Grain Yields in Henan Province during 1961-2013
- Influences of Surface Drip Irrigation on the Growth,Yield and Quality of Several New Species of Guitang Sugarcane
- Fuzzy Analysis Method for Orthogonal Test on Seed Propagation of Eurya chinensis