
二维制备高效液相色谱分离塞隆骨强极性多肽
2015-01-08 08:10陶燕铎王启兰张耀洲梅丽娟
天然产物研究与开发 2015年7期
江 磊 ,赵 换,陶燕铎,王 硕,邵 赟,王启兰,张耀洲,梅丽娟*
1中国科学院西北高原生物研究所,西宁 810001;2 瀚盟生物技术(天津)有限公司;3天津大学药学院,天津 300072;4 中国科学院大学,北京 100049
塞隆骨药材是一种名为高原鼢鼠的啮齿目动物的风干骨骼。塞隆骨药材广泛使用于青藏高原地区。1991 年塞隆骨药材被国家药监局批准成为新中国第一个用于治疗类风湿关节炎等病症的动物类一类新药材,并进入中国药典。随后以塞隆骨单味药制成的塞隆风湿酒取得了中药一类新药的新药批准文号,在2000 例的临床实验中起效率超过90%。除了具备抗类风湿关节炎[1]的功效,塞隆骨还具备抗骨质疏松[2]、止痛[3]和抗炎[4]的功效。由于塞隆骨药材的化学成分过于复杂,几乎没有关于塞隆骨药材化学成分的研究结果发表。为了研究塞隆骨这种复杂动物类药材的化学组成,天然产物研究领域急需开发一种先进的针对动物组织提取物的分离方法。
在前期研究中,塞隆骨的分离工作使用传统柱层析进行。传统方法分离的结果非常不理想。究其原因有二:首先这些传统分离方法都是针对植物提取物设计的,并不适用于动物药的分离纯化。其次这些传统方法由于重复性差、分辨率低和柱效低等缺点,没有能力分离塞隆骨这样的复杂混合物。为了分离塞隆骨混合物,本研究团队不得不寻求更高分离能力的分离方法。
制备型高效液相色谱由于具备高柱效、重复性优异以及高自动化等特点,非常适合并且有能力分离复杂天然产物提取物。在许多天然产物研究中高效液相色谱往往是作为最后一步最精细的纯化方法使用[5]。但是面对复杂的塞隆骨提取液,仅仅使用一维高效液相色谱不能将塞隆骨混合物分离成单体化合物。因此,想要得到塞隆骨单体化合物必须使用多维高效液相色谱。所谓的多维色谱指的是使用多根具备一定正交性的高效液相色谱柱进行配合分离的色谱系统。一旦正交体系形成,多维色谱的分离能力将比一维色谱提升数倍至数十倍。这种具备强分离能力的多维色谱适合用于分离塞隆骨提取物[6-9]。但是,如何选择相互正交的色谱柱、如何控制色谱流速、检测波长、上样量等色谱分离的必须参数并不清楚。为了使用二维色谱分离塞隆骨混合物,本文将建立新型的系统的二维分离方法对塞隆骨提取液进行色谱分离。此方法不仅仅适用于塞隆骨的分离纯化,还可以应用于其他类似的动物药材分离分析。
1 材料与方法
1.1 材料与试剂
实验样品塞隆骨采自青海省海东地区,由中国科学院西北高原生物研究所副研究员高级工程师梅丽娟鉴定为高原鼢鼠骨骼。甲醇、乙腈为天津康科德有限公司提供。
1.2 仪器
制备型高效液相色谱:配备两台100 mL 柱塞杆20 Mpa 高压液相泵、紫外检测器、六通阀进样器和博纳艾吉尔高效液相系统软件(博纳艾吉尔,天津)。
半制备高效液相系统:配备两台50 mL 柱塞杆20 Mpa 高压液相泵、紫外检测器、六通阀进样器和博纳艾吉尔高效液相系统软件(博纳艾吉尔,天津)。
分析型高效液相系统使用日立高效液相系统:配备日立L2130 四元高压泵、日立L2400 紫外检测器、日立L2200 自动进样器和日立Lachrom Elite 高效液相软件(日立,日本)。
质谱系统采取纳升级超高效液相色谱与离子肼质谱(热电,美国)联用的方式(LTQ-XL);质谱原始数据分析使用软件Thermo Proteome Discover 1.2 的SEQUEST 算法(热电,美国)。
2 实验方法
2.1 样品制备
1 kg 新鲜塞隆骨首先在-80 ℃中预冻24 h,然后置入-40 ℃超低温冷冻粉碎机中冷冻粉碎。冷冻粉碎机将塞隆骨原料粉碎至150 目左右后,将冰冻粉末迅速按照1∶20 的料液比置入100 ℃热水中搅拌1 h。加热搅拌结束后将提取液离心取上清液,离心机转速12000 rpm,离心时间40 min。随后将离心上清液置于70 ℃旋转蒸发仪中进行浓缩,并将浓缩液置入4 ℃环境中24 h。将4 ℃的浓缩液过0.45 μm 滤膜后得到色谱样品。此时样品浓度为30.7 mg/mL。
2.2 样品前处理
色谱样品首先使用SPE 固相萃取柱进行前处理。SPE 固相萃取柱柱材料为C18(50 μm,120 Å,日本大曹)。首先用纯甲醇活化SPE 柱,然后用5%甲醇-水(v∶v)溶液平衡SPE 柱后上样。上样后先用5%甲醇-水(v∶v)溶液冲洗SPE 柱,然后用10%甲醇-水(v∶v)溶液洗脱此样品中的大极性组份。收集此大极性组份并将样品浓缩、过膜。过膜后的样品浓度为255.7 mg/mL。此大极性混合物样品即为本文主要研究对象。
2.3 色谱条件
第一维制备色谱在制备型高效液相色谱仪上使用C18AQ 制备色谱柱(50 ×250 mm i.d.,10 μm,120 Å,YMC)。流动相使用水(A)-甲醇(B)体系。线性洗脱方法为:0~15 min,1% B;15~30 min,1%~10% B。流速为80 mL/min。进样量为4 mL。紫外检测波长为214 nm。通过第一维色谱将混合物分成了若干次级组份。每个次级组份用旋转蒸发仪除去溶液后进行称量,再用5%甲醇-水(v∶v)复溶并过0.45 μm 滤膜,制成二维色谱样品。控制复溶溶液的体积使各个样品的浓度为500 mg/mL 左右。
第二维制备色谱在半制备型高效液相色谱仪上使用C18MP 色谱柱(10 ×250 mm i.d.,10 μm,100 Å,Agela)。流动相使用水(A)-乙腈(C)体系。流速为4 mL/min。上样量400 μL。对每个次级组份均采用等度洗脱的洗脱方法,这些洗脱方法分别为:Fr.1(1.1% C 洗脱30 min),Fr.2(1.3% C 洗脱30 min),Fr.3(1.9% C 洗脱30 min)。紫外检测器检测波长为214 nm。
分析型色谱在日立高效液相色谱仪上使用C18MP 色谱柱(5 ×250 mm i.d.,10 μm,100 Å,Agela)。流动相使用水(A)-乙腈(C)体系。流速为1 mL/min。上样量10 μL。对每个化合物均采用等度洗脱的洗脱方法,这些洗脱方法分别为:Fr.1-1(1.1%C 洗脱30 min),Fr.2-1(1.3% C 洗脱30 min),Fr.2-2(1.3% C 洗脱30 min),Fr.2-3(1.3% C 洗脱30 min),Fr.3-1(1.9% C 洗脱30 min)。紫外检测器检测波长为214 nm。
Nano-LC-MS/MS 使用C18色谱柱(0.15 × 100 mm i.d.,1.7 μm,300 Å,Column)。流动相使用0.1%甲酸水溶液(D)-0.1%甲酸乙腈溶液(E)体系。流速为200 μL/min,分流后2μL/min。上样量为20 μL。洗脱方法为:0~15min 5%B,15~60 min 5%B~32%B,60~95min32%~90% B。质谱参数为毛细管电压3.5KV,离子源温度250 ℃,扫描范围100~1800 m/z,肽段碎裂模式为诱导碰撞解离,碰撞能量为35%,质谱数据采集软件为Xcalibur version 1.0.0.2;质谱数据分析软件为Proteome Discoverer 1.2,数据筛选条件是Xcorr >1.9 Charge=1;Xcorr>2.2 Charge=2;Xcorr >3.75 Charge=3;数据库为uniprot 高原鼢鼠蛋白质数据。
3 结果与讨论
3.1 一维制备色谱和固相萃取
塞隆骨水提取物首先在SPE 固相萃取柱上进行预处理。塞隆骨水提取物在SPE 柱上样后先用5%的甲醇溶液冲去大分子杂质,再用10%甲醇溶液洗脱塞隆骨强极性组分。此强极性组分对高分辨率的C18AQ 反向色谱柱来说极性分布依然很广。因此用多重梯度洗脱的方式对样品进行洗脱。最后选择色谱条件为:0~15 min,1% B,15~30 min 1%~10% B;流速为80 mL/min;上样样品浓度为30.7 mg/mL;进样量为4 mL;紫外检测波长为214 nm。此方法制备时间为30 min,冲洗色谱柱与平衡色谱柱的时间一共30 min,每次进样一共花费60 min。如图1 所示,强极性样品被分成了3 个次级组份。这些次级组份经过浓缩干燥后被称重,组份的质量从2 号的0.15 g 到3 号的1.24 g 不等。整个一维色谱制备的回收率为77.3%。
由于本研究的研究对象为大极性小分子多肽,在色谱制备之前首先将样品用固相萃取柱SPE 进行预处理。固相萃取预处理有如下几个好处:(1)塞隆骨水提物粗品的成分非常复杂,其中包含的物质极性分布非常广,用固相萃取柱对其中的大极性物质进行预富集可以得到更符合本研究要求的大极性混合物;(2)塞隆骨水提物中不乏含有大量的两性大分子蛋白质,这些蛋白质在水中溶解度非常好,但是却会在C18色谱柱上死吸附,造成色谱柱柱效和寿命降低。用SPE 预先除掉这类物质可以保护色谱柱;(3)用SPE 固相萃取柱预处理后的样品在溶解性质上极具均一性,可以以非常大的浓度溶解于某一种液态溶剂中,这样就可以最大化地避免因样品体积过大带来的色谱系统分辨率降低的问题。
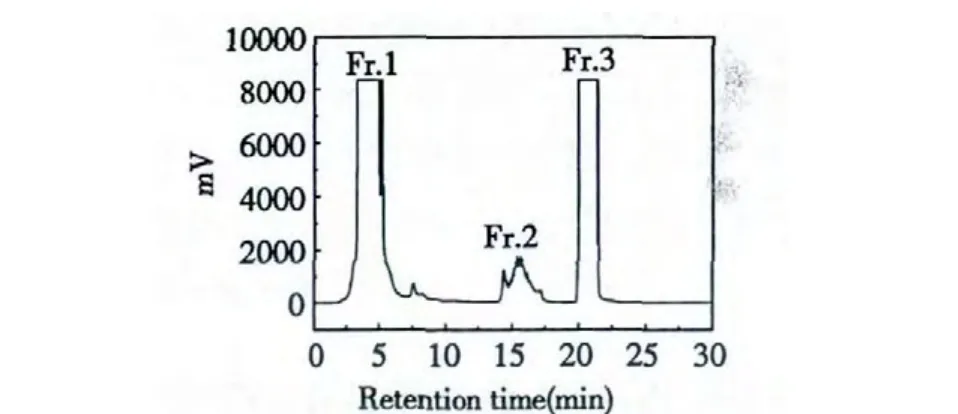
图1 强极性多肽混合物的一维液相制备Fig.1 The first dimensional preparation of strong polar peptides extracts
一维制备色谱实验在C18AQ 制备色谱柱(50 ×250 mm i.d.,10 μm,120 Å,YMC)上进行。选择此色谱柱的原因是分离大极性物质时在反向色谱柱上只能用少量的强洗脱剂才能保证被分离物具有一定保留因子。而反向色谱柱是一种液-液交互作用色谱柱,在工作时被分离化合物在靠近色谱柱基质的液层与流动相之间进行动态交换。对于水-甲醇流动相体系的普通C18反向色谱分离,一旦甲醇在流动相中的比例小于5%,靠近色谱填料基质的液层就会遭到严重破坏,色谱体系分离能力严重下降。所以用普通C18反向色谱柱分离大极性化合物时,即便将流动相中甲醇浓度降至5%以下,也不一定能够增加被分离大极性化合物群的峰容量与分辨率。为了解决这个问题,我们选用在色谱填料基质的C18基团之间键合少量极性基团的极性共聚C18色谱柱。这种新型C18反向色谱柱能在更低的甲醇浓度(小于1%)下稳定工作,也能明显增强强极性化合物的保留因子,非常符合本研究的要求。
3.2 次级组份的二维制备
色谱分离方法的分离能力是衡量色谱系统的主要指标。这个指标最重要的一个评价依据是分离系统的峰容量。峰容量是指色谱系统第一个峰与最后一个峰之间具备一定分辨率的峰个数。峰容量与分离材料的种类、分离方法、流动相、被分离物与分离设备等因素息息相关。一维高效液相色谱的峰容量相比传统柱层析、薄层色谱与排阻色谱有了飞跃性的提高,但是还不能满足分离如同塞隆骨这样极复杂天然产物的要求。虽然在一维色谱的基础上通过改变柱长、柱温、流动相配比和柱材料可以提高峰容量,但是这种提高非常有限,若想大幅度提高色谱系统的峰容量达到分离复杂天然产物的要求,就必须使用多维色谱。多维色谱峰容量是指所使用所有的单一色谱分离系统的峰容量的乘积。如果一维色谱的峰容量达到500,二维色谱即便使用20 的峰容量,总体系统的峰容量也能提高20 倍,达到10000。这样就可以轻松对复杂天然产物混合物进行分离了[10-13]。
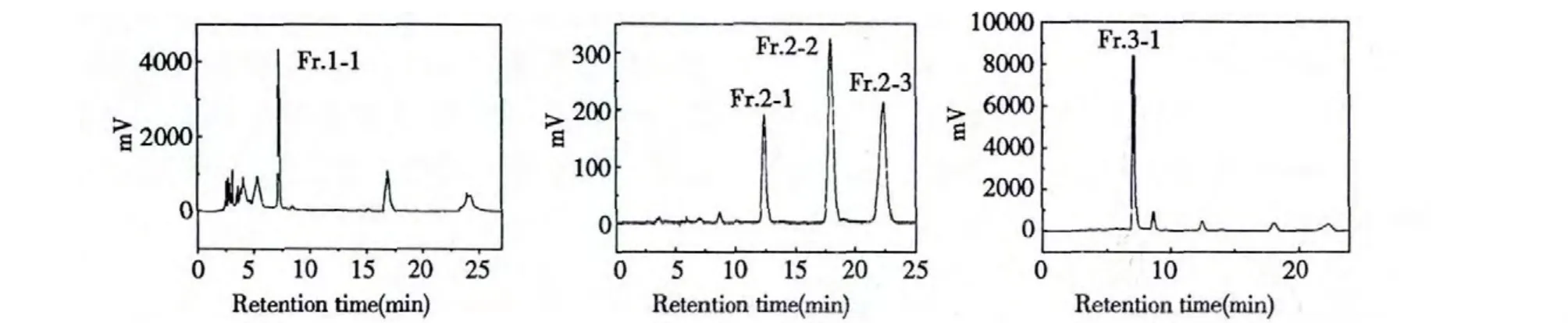
图2 次级组分的二维液相制备Fig.2 The second dimensional preparation of the second fractions
由图1 可知,虽然色谱样品在SPE 柱上是5%甲醇至10%甲醇中间的一段,其组成依然比较复杂,在色谱图上只能分辨出三个主要的峰群,这三个峰群中还包含若干单体化合物峰。在一维色谱中虽然已经将流动相的甲醇浓度降至1%,整体峰分布在0~30 min 之内,依然得不到单体化合物峰。这说明此混合物的组成过于复杂,已经远超一维色谱可以分离的复杂范围,若要想将此混合物分离成单体化合物,就必须使用二维色谱。Fr.1、Fr.2 和Fr.3 的二维色谱方法首先在相同填料的分析柱上进行摸索,然后放大到制备柱上来由于此时次级组份中的物质性质已经非常相似,用等度洗脱程序就可以达到对其中化合物分离的目的。Fr.1、Fr.2 和Fr.3的二维分离色谱图见图2 所示:Fr.1 的组成过于复杂,在二维色谱上仅仅分离出一个主要的单峰,其余物质在色谱图上都是含量接近的低矮碎峰。Fr.2和Fr.3 的二维分离较为成功,均分离成了若干单体化合物。通过对次级组份单体化合物的制备,得到了Fr.1-1(2.2 mg)、Fr.2-1(0.4 mg)、Fr.2-2(0.6 mg)、Fr.2-3(0.5 mg)和Fr.3-1(1.7 mg)五个具备足够进行结构鉴定所必需质量的化合物。
3.3 所分离的小分子肽类化合物的结构鉴定
分离得到的Fr.1-1、Fr.2-1、Fr.2-2、Fr.2-3 和Fr.3-1 五个化合物用基于质谱系统的多肽分析方法进行序列鉴定。经过Nano-LC-MS/MS 鉴定得到五个化合物的二级质谱,利用Thermo proteome Discover 1.2 的SEQUEST 算法与高原鼢鼠蛋白质数据库比对,同时结合denovo 测序推断得到这五个肽段序列如表1 所示。
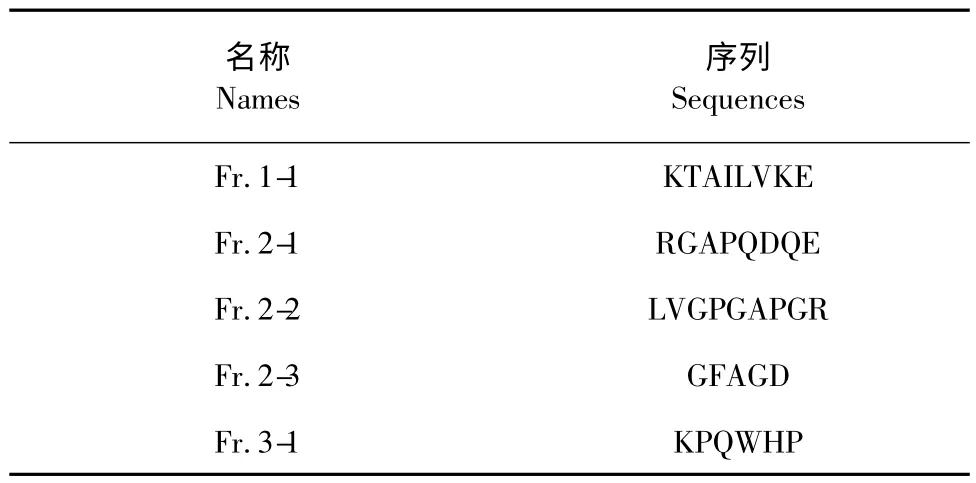
表1 分离出的小分子多肽序列Table 1 The sequences of the purified peptides
4 结论
本研究建立了一种针对动物组织高极性肽段的分离方法。首先利用SPE 固相萃取柱对粗样品进行预处理,截取其中的高极性组份作为本研究的研究对象;然后使用亲水性的C18AQ 制备柱将SPE 柱后的高极性组份分成了若干次级组份,这些次级组份是一类具备相似结构的简单混合物;最后利用C18MP 色谱柱将次级组份分离成单体化合物。得益于二维液相色谱的高峰容量和C18AQ 色谱柱与C18MP 色谱柱之间的正交性,分离单体化合物时均得到了高分辨率的色谱图。此方法适用于所有复杂肽类化合物的分离,不仅仅可以得到高纯度单体化合物,还具备很高的制备效率。推广此方法将会对天然肽类药物研究起到很好的推动作用。
1 Zhao XH(赵晓辉),Jiang FQ (将福泉),Yue HL (岳会兰),et al.The pharmacodynamics study of Osteon Myospalacem Baileyi extract on experimental rheumatoid arthritis rat model.Chin Tradit Pat Med(中成药),2007,29:1221-1223.
2 Hai P (海平).Study on Osteon Myospalacem Baileyi bone in treating osteoporotic rats.Shandong J Tradit Chin Med(山东中医杂志),2002,21:231-233.
3 Hai P (海平).The analgesic activity of Osteon Myospalacem Baileyi and its effect on single amine neurotransmitter in the brain.Shandong J Tradit Chin Med(山东中医杂志),2001,20:232-235.
4 Hai P (海平).The anti-inflammatory effects of Osteon Myospalacem Baileyi extract.Liaoning J Tradit Chin Med(辽宁中医杂志),2000,27:524-526.
5 Han Z,Liu X,Ren Y,et al.A rapid method with ultra-highperformance liquid chromatography-tandem mass spectrome-try for simultaneous determination of five type B trichothecenes in traditional Chinese medicines.J Sep Sci,2010,33:1923-1932.
6 Giddings J.Maximum number of components resolvable by gel filtration and other elution chromatographic methods.Anal Chem,1967,39:1027-1028.
7 Giddings J.Concepts and comparisons in multidimensional separation.Hrc-J High Res Chrom,1987,10:219-323.
8 Giddings J.Two-dimensional separation:concept and promise.Anal Chem,1984,56:1258A-1270A.
9 Jandera P.Column selectivity for two dimensional liquid chromatography.J Sep Sci,2006,29:1763-1783.
10 Horie K,Kimura H,Ikegami T,et al.Calculating optimal modulation periods to maximize the peak capacity in two-dimensional HPLC.Anal Chem,2007,79:3764-3770.
11 Davis JM.Effect of first-dimension under sampling on effective peak capacity in comprehensive two-dimensional separations.Anal Chem,2008,80:461-473.
12 Li X,Stroll D,Carr P.Equation for peak capacity estimation in two-dimensional liquid chromatography.Anal Chem,2009,81:845-850.
13 Liu Z,Patterson J.Geometric approach to factor analysis for the estimation of orthogonality and practical peak capacity in comprehensive two dimensional separation.Anal Chem,1995,67:3840-3845.
猜你喜欢
传染病信息(2022年6期)2023-01-12
纺织标准与质量(2022年3期)2022-08-10
承德医学院学报(2022年2期)2022-05-23
建材发展导向(2021年13期)2021-07-28
时代英语·高一(2019年5期)2019-09-03
天然产物研究与开发(2018年9期)2018-10-08
成长·读写月刊(2017年8期)2017-08-12
中国洗涤用品工业(2015年9期)2015-02-28
航天返回与遥感(2014年1期)2014-07-31
郑州大学学报(工学版)(2014年6期)2014-03-01
