
分散液液微萃取-反相液液微萃取-扫集-胶束电动色谱法测定红酒中的3种氯酚类物质
2014-12-24 03:51孙建芝刘书慧
色谱 2014年3期
孙建芝, 贺 晖, 刘书慧,2*
(1.西北农林科技大学理学院,陕西 杨凌712100;2.旱区作物逆境生物学国家重点实验室,陕西 杨凌712100)
氯酚类物质被广泛用作漂白剂、杀菌剂、杀虫剂和木材防腐剂等。近年来在废弃物和污泥[1]、土壤[2]、地下水及地表水[3]等中均已检测到氯酚类污染物,甚至有文献[4,5]报道在红酒中也检测到痕量氯酚类物质。氯酚具有毒性大、难生物降解、“三致”(致癌、致畸、致突变)效应等危害,已被美国环保局(EPA)[6]和我国国家环保总局[7]列入优先控制污染物的黑名单。因此,建立一种简单、快速、灵敏的氯酚类物质测定方法已成为亟待完成的任务。
氯酚类物质在红酒中的含量很低,传统的分析方法难以得到可靠结果。目前,氯酚类化合物的提取方法主要有液液萃取法[8]、固相萃取法[9]、固相微萃取法[10]、顶空固相微萃取法[11]等。这些方法在样品前处理中的应用已比较成熟,但是存在有机溶剂消耗大、操作繁琐、装置昂贵等缺点。分散液液微萃取(DLLME)是2006年由Rezaee等[12]率先提出的一种萃取富集技术。该方法具有操作简单、快速、费用低、富集倍数高、萃取效果好和对环境友好等优点,因此已被广泛使用。近几年,基于DLLME富集方法,新颖的反相分散液液微萃取技术(RPDLLME)[13,14]也得到广泛应用。
DLLME 常与气相色谱-质谱/电子捕获检测(GC-MS/ECD)[4,5]和 高 效 液 相 色 谱-紫 外 检 测(HPLC-UV)[15,16]联用测定氯酚类物质。Campillo等[4]通过对分析物衍生化建立了DLLME-GC-MS模型,红酒和软木塞中的待分析物经乙酸酐衍生后用200μL CCl4萃取,最后取4μL萃取相进行GC分析测定。Pizarro等[5]使用CCl4作萃取剂,测定了葡萄酒中的卤代酚和卤代苯甲醚等物质。Hadjmohammadi等[15]建 立 了 乙 醇 辅 助 萃 取(AA)-DLLME-HPLC-UV 测定方法,并测定了3 种水样中五氯酚的含量。这些方法中,GC 需要将样品衍生化,操作过程繁琐、冗长,而且使用的萃取剂均为毒性较大的含氯萃取剂;HPLC 灵敏度低,而且使用甲醇或乙腈作为流动相,有机溶剂消耗多,污染严重。
毛细管电泳(CE)因其分离效率高、分析速度快、有机溶剂消耗少等优点在环境分析和食品分析中广泛应用,但CE-UV 灵敏度较低,仍是制约其发展的一个难题。如何提高检测灵敏度以满足检测复杂样品中痕量和超痕量组分的要求,依然是电泳研究领域中的焦点与热点。在线富集技术是样品进样或者分离过程中提高灵敏度的方法,常用的有场强放大[17]、动态pH 连接[18]、扫集[19]和等电聚焦[20]等技术。近年来,已有报道将DLLME 与上述电泳在线富集方法联用,如Us等[17]利用DLLME-fieldamplified sample injection(FASI)联用建立了测定牛尿中β2-兴奋剂类物质的方法,该方法的LOD 为1.8~37.0μg/L,富集倍数为41~1 046,4种分析物 在5.5 min 内 被 检 测。Zhang 等[19]建 立 了DLLME-扫集-MEKC 方法测定黄瓜中的4 种新烟碱类杀虫剂,该方法具有富集倍数高、重现性好和回收率高等优点。基于上述DLLME 与CE 联用的优点,本研究选取五氯酚(PCP)、2,4,6-三氯酚(TCP)和2,4-二氯酚(DCP)等3种氯酚为模型分析物,建立了DLLME和RP-LLME两步萃取与扫集-MEKC联用的分离富集方法,并将该方法成功应用于市售红酒中氯酚类物质的分析检测。
1 实验部分
1.1 仪器、试剂与材料
P/ACETMMDQ 型毛细管电泳仪(美国Beckman Coulter公司),配备二极管阵列检测器;弹性石英毛细管(50.2 cm×75μm,有效长度40 cm,河北永年锐沣色谱器件有限公司);Direct-Q3型纯水仪(美国Millipore公司);Allegra X-12型离心机(美国Beckman Coulter公司)。
PCP、TCP 和DCP 标 准 品(纯 度 为98%~99%,阿拉丁试剂公司);离子液体[C4MIM][PF6](C4)、[C6MIM][PF6](C6)和[C8MIM][PF6](C8)(成捷化学有限公司);正辛醇、磷酸三丁酯均为分析纯(阿拉丁试剂公司);正己烷、乙腈和甲醇均为色谱纯;磷酸二氢钠、盐酸、氯化钠、氢氧化钠、磷酸均为分析纯;十二烷基硫酸钠(SDS)(美国Sanland 公司)。实验所用的11 种不同品牌红酒(酒精含量11.5%~12%(v/v))均购于本地超市,并将样品依次编号为1~11号。
PCP标准储备液(0.2 g/L)、TCP和DCP标准储备液(2.0 g/L)均用甲醇配制于10 mL棕色容量瓶中,置于-10 ℃下避光保存。实验时不同浓度的工作溶液由标准储备液稀释而得。
1.2 电泳过程
新毛细管实验前需活化,活化步骤同文献[21]所述,其中冲洗压力均为137.8 kPa。电泳缓冲溶液为25 mmol/L NaH2PO4-100 mmol/L SDS-30%(v/v)乙腈,pH2.3;分离电压为-15 kV;检测波长为214 nm;样品基体为80 mmol/L NaH2PO4,压力进样(20.67 kPa×20 s)。
不同浓度的电泳缓冲溶液由200 mmol/L NaH2PO4和400 mmol/L SDS 储 备 液 制 备,3 mol/L H3PO4溶液调节pH 值,缓冲溶液使用前用0.22μm 滤膜过滤并超声脱气5 min。
1.3 实际样品处理
1.3.1 分散液液微萃取过程
取3.5 mL红酒(浓HCl调节pH 至3.0;加入固体NaCl至120 g/L)置于一个具有细长颈的一次性聚乙烯萃取管中,加入300μL 正己烷,轻摇3 min后离心(5 000 r/min,3 min),上层正己烷相用微量进样器准确吸取并转移到1.5 mL离心管中。
1.3.2 反相液液微萃取过程
取25μL 0.16 mol/L NaOH 溶液加入到上述正 己 烷 相 中,轻 摇2 min 后 离 心(5 000 r/min,2 min),用微量进样器准确量取沉积于离心管底部的NaOH 溶液(25±1)μL。然后加入(25±1)μL 0.16 mol/L H3PO4溶液中和NaOH,离心(12 000 r/min,2 min)后直接进行电泳分离富集测定。
2 结果与讨论
2.1 电泳参数的优化
2.1.1 运行缓冲溶液的选择
在扫集-胶束电动色谱富集法中,为了抑制电渗流,使SDS的电泳速度大于电渗流速度,选用酸性磷酸盐缓冲溶液(pH2.3)。缓冲溶液体系中添加有机添加剂可以调节分析物在胶束和缓冲体系两相中的分配能力,从而改善分析物的分离度和峰形。常用的有机添加剂有乙腈、乙醇、甲醇和异丙醇等。实验结果表明,缓冲溶液中加入30%(v/v)乙腈时样品中的分析物达到完全分离且几乎没有杂质干扰。考虑分析物的分离度和迁移时间,故缓冲溶液中加入30%(v/v)乙腈。
实验考察了NaH2PO4缓冲液(pH2.3)浓度在15~60 mmol/L 范围内变化时对分析物分离度和灵敏度的影响。随着缓冲液浓度的增加,迁移时间逐渐缩短,当NaH2PO4浓度为25 mmol/L 时分析物的峰高达到最大。因此选取25 mmol/L NaH2PO4为优化值。
SDS浓度是胶束扫集法中一个重要的参数[22],对氯酚的分离和富集起着重要作用。通过增加SDS的浓度,可以提高电泳运行缓冲液溶解样品的能力,也有利于样品的富集。但随着SDS浓度的增大,SDS扫过样品区带后穿过缓冲溶液界面的速度会增大,导致样品区带展宽,甚至峰高降低。同时,随着SDS浓度的增加,电泳电流及焦耳热也会增大,体系温度会升高,出峰稳定性也受到消极影响。本实验考察了60~140 mmol/L 范围内不同浓度SDS对氯酚分离和富集效果的影响。结果表明,随着SDS浓度的增加,分析物的峰高增加;当SDS浓度大于100 mmol/L时,分析物峰高基本不再增加,而且峰展宽,不利于分离测定。因此选择100 mmol/L SDS为最优值。
2.1.2 样品基体组成和进样时间的选择
考察了样品基体中NaH2PO4浓度(20、50、80和100 mmol/L)对分析物富集倍数的影响,结果见图1。随着NaH2PO4浓度的增加,3种物质的迁移时间逐渐缩短,灵敏度增加;但是当NaH2PO4浓度大于80 mmol/L时,分析物的灵敏度不再增加。因此选择80 mmol/L NaH2PO4作为样品基体。
进样时间影响方法的分离度和扫集的富集水平。实验中,考察了20.67 kPa下的3 个进样时间(20、30、40 s),相应的进样长度为17.35、26.03、35.71 cm。如图2所示,分析物的灵敏度随进样时间的增加而增大。当进样时间>30 s时,峰展宽的程度大于峰高增加的程度,不利于实际样品的测定。并且在分析红酒样品的预实验中发现,当进样时间大于20 s时,分析物(尤其是TCP 和DCP)不能与红酒中的杂质达到基线分离,故选择样品进样时间为20 s。
2.2 样品前处理条件的优化
2.2.1 分散液液微萃取
市售红酒中乙醇含量约为12%(v/v),而乙醇与本文选用的萃取剂均能互溶,因此红酒中的乙醇可以作为本实验的分散剂。此处主要考察DLLME过程中萃取剂种类及体积、样品pH 值、离子强度和萃取时间等参数对3种氯酚萃取率的影响。
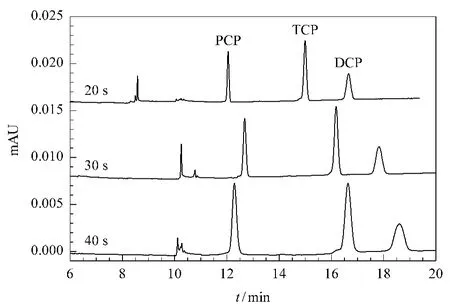
图2 样品进样时间对3种氯酚类物质富集的影响Fig.2 Effect of the injection time on the sweeping efficiency for the three CPs
分散液液微萃取建立于三相溶剂体系,在分散剂的作用下萃取剂以微小液滴的形式分散在样品溶液中,利用化合物在萃取剂和样品溶液中的溶解度或分配系数不同,使化合物从样品溶液中转移到萃取剂中的方法。本文选用多种极性不同的萃取剂进行比较,为方便归类,将萃取剂按密度大小分为两类:密度大于样品溶液的萃取剂C4、C6和C8和密度小于样品溶液的正己烷、正辛醇和磷酸三丁酯。取3.5 mL加标红酒和上述萃取剂各100μL,按1.3节实验步骤考察萃取剂对分析物萃取率的影响,结果如图3 所示。除正己烷外,其他几种萃取剂对PCP和TCP的萃取率很低,尤其是PCP 的萃取率几乎为零。然而已有文献[15,23,24]报道使用正辛醇、磷酸三丁酯和离子液体萃取氯酚,其回收率几乎都满足要求。因此以上实验结果可能是由于第二步RP-LLME引起的。Li等[25]也曾报道碱溶液几乎不能将氯酚尤其是PCP 和TCP 从正辛醇中萃取出来。其可能的原因如下:萃取剂结构的不同,使萃取剂和氯酚之间存在分子间作用力、氢键和π-阳离子作用力等不同作用力。氯酚的羟基与正辛醇和磷酸三丁酯的氧原子可能形成作用力较强的氢键。氯酚与离子液体之间,除了离子液体中阴离子PF6-的F原子与氯酚的羟基可能形成氢键外,离子液体的阳离子与氯酚的苯环间也可能形成π-阳离子作用力,这些强的作用力使氯酚易被这些萃取剂从样品溶液中萃取出来,而不易反萃取到碱溶液中。但是正己烷与氯酚之间只有分子间作用力,不存在氢键和π-阳离子作用力等较强的作用力,所以反萃取效果最优。最后,选择正己烷作为本实验的萃取剂。同时考察了萃取剂体积对萃取效率的影响,结果如图4所示。当正己烷加入量>300μL时,萃取率不再增加,所以选择萃取剂的体积为300μL。
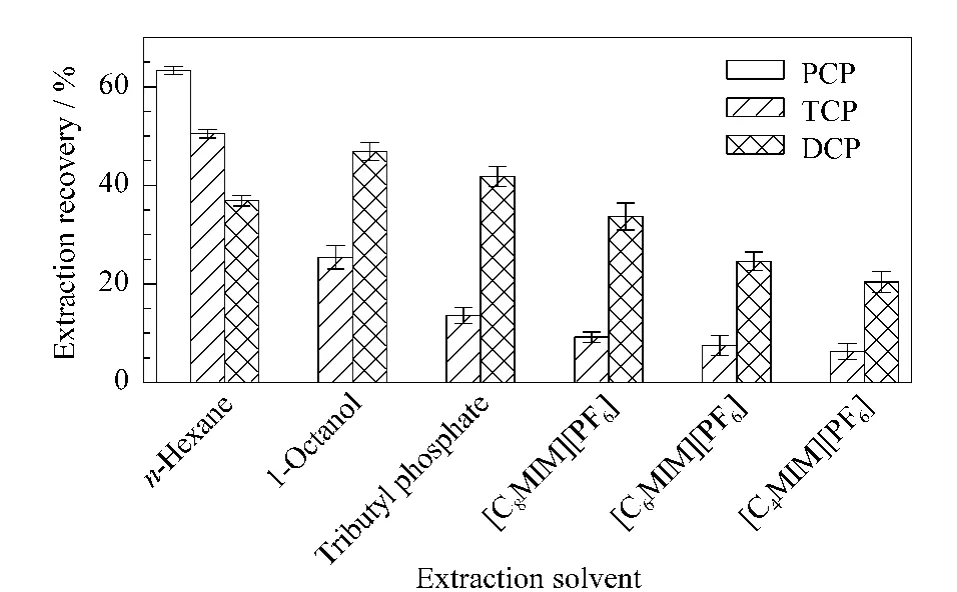
图3 不同萃取剂对3种氯酚萃取回收率的影响(n=3)Fig.3 Effect of the type of the extraction solvent on the extraction recoveries for the three CPs(n=3)
3 种氯酚在25 ℃下 的p Ka分别为4.7、6.0、7.5[26],均为弱酸性物质,因此调节样品的pH 值使其为酸性可使分析物的解离平衡向生成分子方向移动,弱酸性分析物的解离度降低,从而降低分析物在溶液中的溶解度,有利于分析物被有机萃取剂有效萃取。实验结果表明,样品pH 为3.0 时分析物的萃取率最高。
通过加入不同质量的固体NaCl(至0~250 g/L)来考察离子强度对分析物萃取率的影响。结果当NaCl质量浓度为120 g/L时,分析物的萃取率达到最大,随后基本保持恒定。原因可能是随着离子强度的增加,分析物在水中的溶解度减小,从而提高了萃取率;当到达最优时,萃取率就不再变化。经实验得出最优的萃取时间为3 min、离心条件为5 000 r/min下3 min。
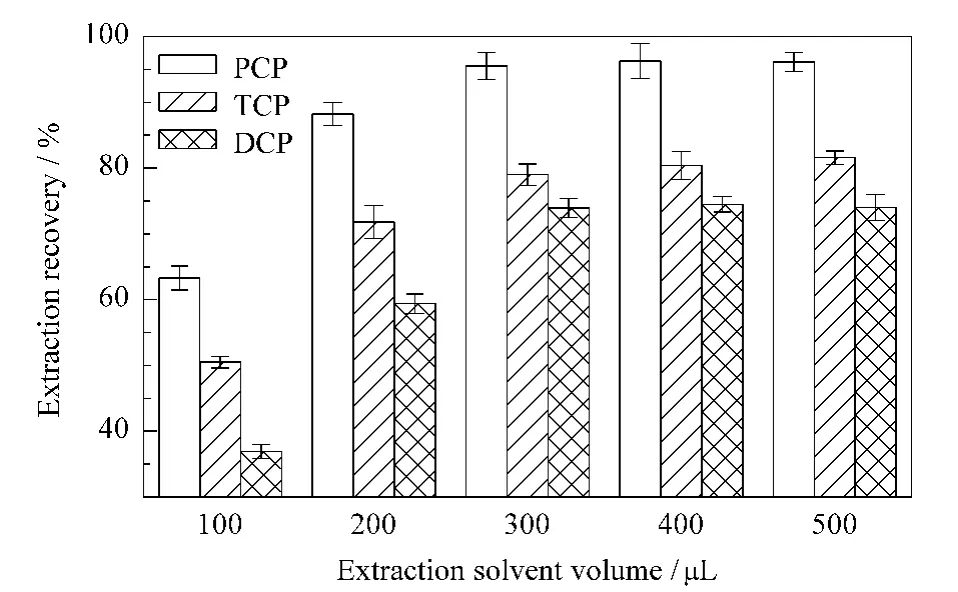
图4 正己烷体积对3种氯酚萃取回收率的影响(n=3)Fig.4 Effect of the volume of hexane on the extraction recoveries for the three CPs(n=3)
2.2.2 反相液液微萃取
反相萃取参数影响分析物的萃取率和富集倍数,同时也影响电泳分析的分离效率和灵敏度。本实验考察了反相萃取过程中NaOH 浓度和体积、萃取时间和离心条件等实验参数对分析物萃取率的影响。
固定NaOH 体积为60 μL,考察不同浓度NaOH 溶液对反相萃取效率的影响,结果如图5a所示。NaOH 的浓度为0.16 mol/L时,分析物的萃取率达到最优。固定NaOH 浓度为0.16 mol/L,不同体积的NaOH 溶液对萃取率的影响如图5b所示。当NaOH 溶液体积大于25μL 时,萃取率不再增加。综合考虑NaOH 溶液体积对分析物萃取效率和富集倍数的影响,选择NaOH 的体积为25μL。
另外,通过优化发现萃取时间为2 min、离心条件为5 000 r/min下2 min时萃取率即可达到最高。
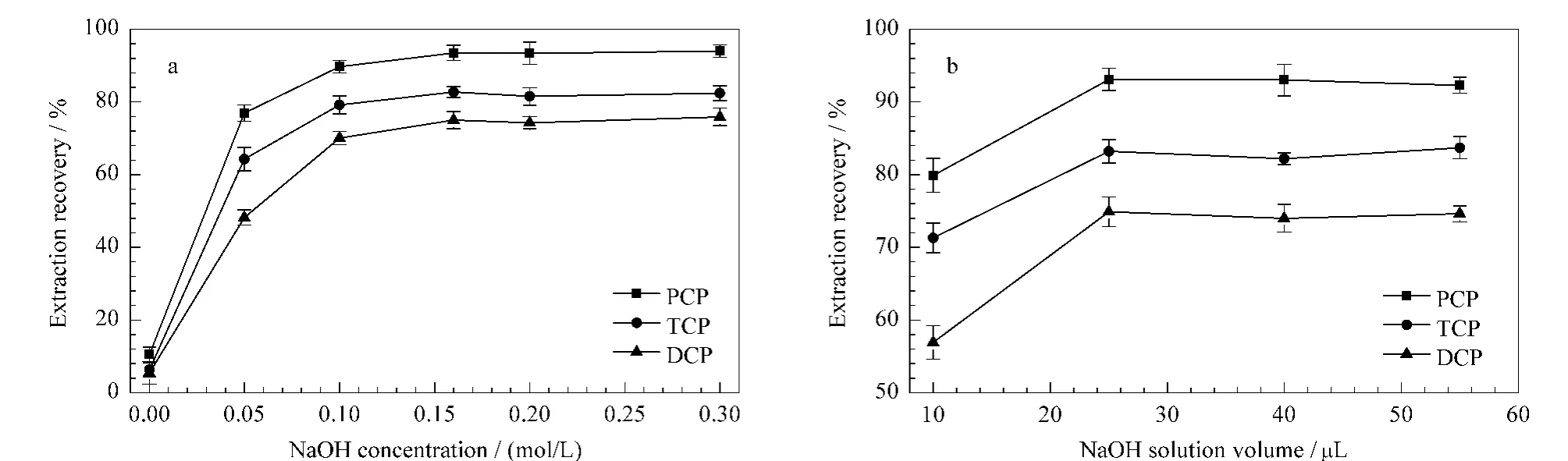
图5 NaOH 溶液(a)浓度和(b)体积对3种氯酚萃取回收率的影响(n=3)Fig.5 Effect of the(a)concentration and(b)volume of NaOH solution on the extraction recoveries for the three CPs(n=3)
2.3 线性范围、检出限、定量限和富集倍数
在优化的富集、分离条件下分析了10个不同质量浓度(0.5~100μg/L)的空白样品加标溶液,考察了各组分的线性范围、检出限(S/N =3)、定量限(S/N=10)和富集倍数,结果如表1所示。
PCP和TCP的线性范围为0.5~100μg/L(r≥0.991 0),DCP的线性范围为1.5~80μg/L(r=0.985 1)。比较文献[27,28]报道的其他CE-UV 方法所得的氯酚类物质的检出限(3.5~100μg/L),本方法的检测灵敏度高2~3个数量级。灵敏度虽不及GC-MS/ECD[4,5],但 其 富 集 倍 数 远 远 大 于GCMS/ECD。
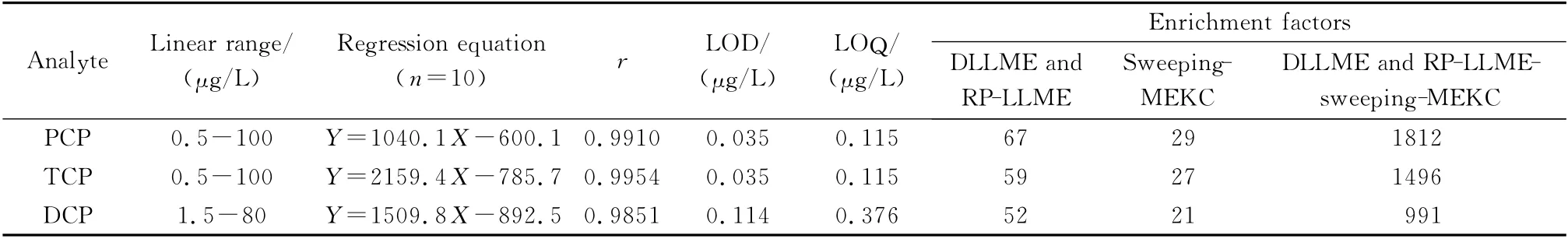
表1 3种氯酚类物质的线性范围、回归方程、检出限、定量限和富集倍数Table 1 Linear ranges,regression equations,LODs,LOQs and enrichment factors for the three CPs
2.4 方法的回收率及精密度
考察2个样品(1号、2号)在3个加标水平下的回收率和精密度,按优化的实验条件对每个加标水平进行3次平行实验,结果如表2所示。3 种物质的平均回收率为75.2%~104.7%,且相对标准偏差≤6.17%。
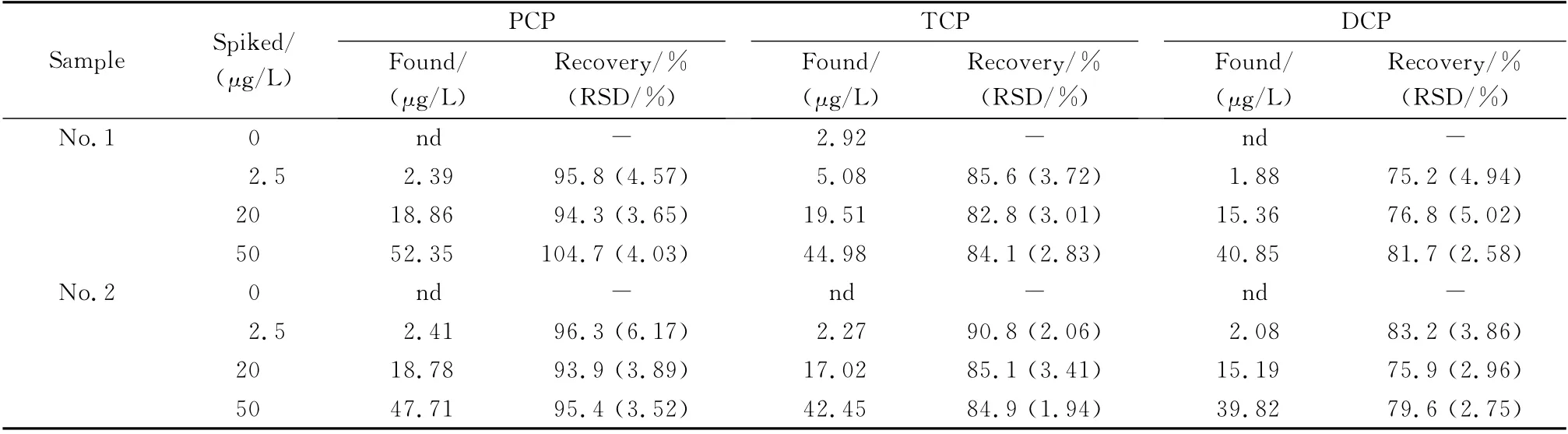
表2 两个红酒样品中3种氯酚类物质的加标回收率(n=3)Table 2 Recoveries of three CPs spiked in two red wine samples(n=3)
2.5 实际样品的测定
在优化的实验条件下对市售11种红酒进行了分析,检出1号、3号、8号和11号红酒样品中含有TCP,含量分别为2.92、1.13、0.61 和1.75μg/L,所有样品中均检测不到PCP和DCP。目前没有相关标准规定红酒中氯酚的最高限量,根据我国生活饮用水卫生标准[29]中对氯酚类物质的限量标准得出这些红酒中的TCP含量没有超标。部分实际样品的电泳谱图见图6,可以看出该方法在16 min内即可检测出3种氯酚,并且几乎没有杂质干扰。
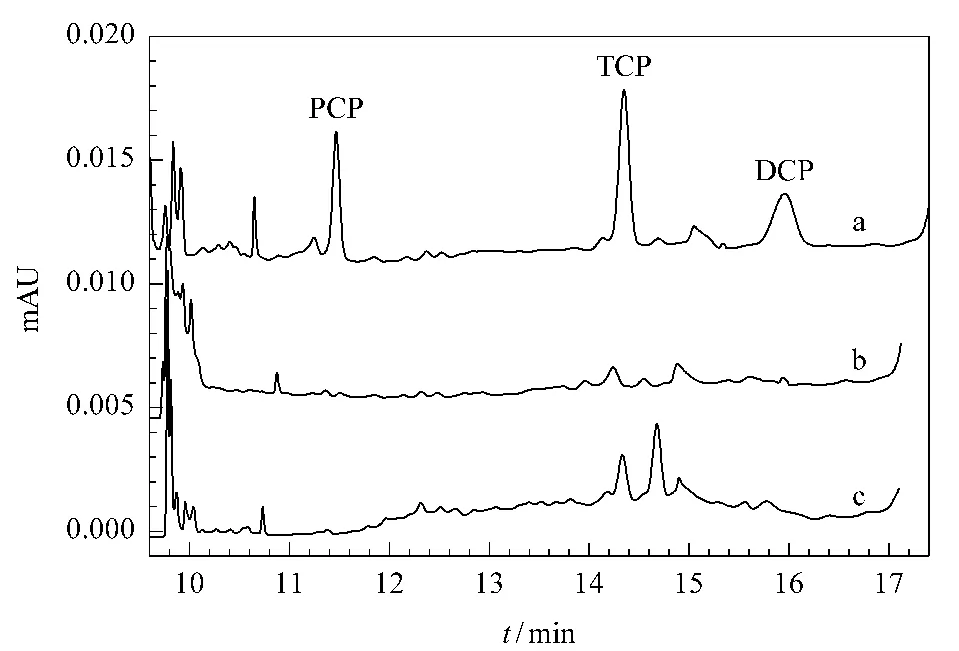
图6 (a)3号红酒加标(3种氯酚各15μg/L)样品、(b)3号红酒样品和(c)1号红酒样品的电泳谱图Fig.6 Electropherograms of(a)No.3 red wine sample spiked with the three CPs(15μg/L);(b)No.3 red wine sample and(c)No.1 red wine sample
3 结论
本实验系统考察了影响扫集-胶束毛细管电动色谱富集技术中富集效率和分离度的各种因素,同时考察了样品前处理(DLLME 和RP-LLME)过程中影响分析物萃取率的各种参数。通过优化这些参数,建立了离线富集-在线富集联用技术检测红酒中3种氯酚类物质的分析方法.该方法具有富集倍数高、灵敏度高、简单易行、重现性好、成本低等优点,成功应用于市售11种红酒中氯酚的测定。红酒有促进睡眠、降低疾病发生率、美容养颜和改善血液循环等优点,越来越多的人选择饮用红酒。而本实验中检测出某些品牌红酒中含有痕量的氯酚,因此红酒的质量安全问题也应给予关注。
[1] Morales S,Canosa P,Rodriguez I,et al.J Chromatogr A,2005,1082(2):128
[2] Padilla-Sanchez J A,Plaza-Bolanos P,Romero-Gonzalez R,et al.J Chromatogr A,2010,1217(14):5724
[3] Gao X J,Zhang Y,Sun S P.Chinese Journal of Health Laboratory Technology(高学杰,张毅,孙仕萍.中国卫生检验杂质),2011,21(5):1098
[4] Campillo N,Vinas P,Cacho J I,et al.J Chromatogr A,2010,1217(47):7323
[5] Pizarro C,Saenz-Gonzalez C,Perez-del-Notario N,et al.J Chromatogr A,2010,1217(49):7630
[6] Rodriguez I,Cela R.Trends Anal Chem,1997,16(8):463
[7] Ying L C,Ouyang W H.Industrial Water &Wastewater(应立春,欧阳文华.工业用水与废水),2013,44(4):86
[8] Minuti L,Pellegrino R M,Tesei I.J Chromatogr A,2006,1114(2):263
[9] Li H Y,Zhang Q,Kang S Y,et al.Chinese Journal of Chromatography(李海玉,张庆,康苏媛,等.色谱),2012,30(6):596
[10] Ho T T,Chen C Y,Li Z G,et al.Anal Chim Acta,2012,712:72
[11] Holopainen S,Luukkonen V,Nousiainen M,et al.Talanta,2013,114:176
[12] Rezaee M,Assadi Y,Hosseini M M,et al.J Chromatogr A,2006,1116(1/2):1
[13] Hashemi P,Raeisi F,Ghiasvand A R,et al.Talanta,2010,80(5):1926
[14] Xiao C Q,Tang M Q,Li J,et al.J Chromatogr B,2013,931(15):111
[15] Hadjmohammadi M R,Fatemi M H,Shakeri P.J Sep Sci,2012,35(1):3375
[16] Moradi M,Yamini Y,Esrafili A,et al.Talanta,2010,82(5):1864
[17] Us M F,Alshana U,Lubbad I,et al.Electrophoresis,2013,34(6):854
[18] Monton M R N,Imami K,Nakanishi M,et al.J Chromatogr A,2005,1079(1/2):266
[19] Zhang S H,Yang X M,Yin X F,et al.Food Chem,2013,133(2):544
[20] Liu S Q,Wang H L.Chinese Journal of Chromatography(刘胜权,汪海林.色谱),2011,29(9):816
[21] Lu Y C,Wang H Y,Song P P,et al.Chinese Journal of Chromatography(卢玉超,王海燕,宋萍萍,等.色谱),2011,29(11):1122
[22] Dawod M,Breadmore M C,Guijt R M,et al.J Chromatogr A,2010,1217(3):386
[23] Li D L,Guo Y Y,Chang Z X,et al.J Chem Eng Data,2013 58(3):731
[24] Galan-Cano F,Lucena R,Cardenas S,et al.J Chromatogr A,2012,1229(16):48
[25] Li X Y,Xue A F,Chen H,et al.J Chromatogr A,2013,1280(1):9
[26] Morais P D,Stoichev T,Basto M P,et al.Talanta,2012,89(1):1
[27] Zhou C H,Tong S S,Chang Y X,et al.Electrophoresis,2012,33(8):1331
[28] Liang T T,Lv Z H,Jiang T F,et al.Electrophoresis,2013,34(3):345
[29] GB5749-2006
猜你喜欢
装备制造技术(2019年12期)2019-12-25
分析化学(2019年3期)2019-03-30
天然产物研究与开发(2018年9期)2018-10-08
中成药(2017年7期)2017-11-22
中成药(2017年3期)2017-05-17
上海建材(2017年4期)2017-04-06
现代检验医学杂志(2016年1期)2016-11-12
上海大学学报(自然科学版)(2016年6期)2016-02-28
山西大同大学学报(自然科学版)(2016年6期)2016-01-30
岩矿测试(2015年3期)2015-12-21
