
杂色龙虾线粒体基因组全序列的测定与分析
2014-12-02 03:10刘海情刘楚吾
海洋科学 2014年11期
刘 皓, 姚 维, 刘海情, 刘楚吾, 刘 丽
(广东海洋大学 水产学院 南海水产经济动物增养殖广东普通高校重点实验室, 广东 湛江 524025)
动物线粒体基因组(mitochondrial DNA, mtDNA)是核外遗传物质, 结构简单, 具有典型的母系遗传特征[1], 受精时, 父本 mtDNA 无法进入卵细胞, 受精卵的 mtDNA直接来自母本, 不存在父母双方mtDNA的重组过程。mtDNA进化速度相对较快, 大约是单拷贝核基因进化速度的6~7倍[2-3]。mtDNA还缺少具有校对功能的酶, 呈游离裸露状态且无组蛋白的保护, 由于其修复系统不健全、代增时间比较短和选择压力小等原因, 造成的突变很容易固定下来。尽管动物线粒体在基因组成上具有很高的保守性,但在种群内或不同物种间存在较大的差异[2,4-6]。利用这种高效的单倍体母系遗传方式以及差异性,只需少量材料就能反映群体的遗传结构, 可缩减用于测有效种群的群体大小, 提高对遗传漂变的敏感性, 便于进行群体分析[7]。后生动物 mtDNA为典型的环状双链分子结构, 长度大多在14~18 kb,编码 37个基因, 包括 13个蛋白质基因、22个tRNA基因以及12s rRNA和16s rRNA基因[8]。近年来, 研究者们非常关注后生动物 mtDNA的基因排列, 主要是因为它是研究系统进化的一个很好的模型[9-12]。
作者报道了杂色龙虾的线粒体基因组全序列特征, 并在杂色龙虾(Panulirus versicolor)、中国龙虾(Panulirus stimposni)、波纹龙虾(Panulirus homarus)3个物种线粒体基因组全序列的基础上分析了龙虾属线粒体基因组多态位点及遗传变异, 为进一步从分子水平对龙虾类的分类研究奠定基础。
1 材料与方法
1.1 实验材料
实验所用杂色龙虾样品来自广东湛江霞山水产品市场, 取其肌肉, –20℃保存于无水乙醇中备用。
1.2 基因组总DNA的提取
以杂色龙虾的肌肉为实验材料, 参照《现代分子生物学实验技术》, 采用苯酚-氯仿法提取基因组总DNA[13]。并利用核酸定量仪检测DNA的浓度和纯度,然后4℃保存备用。
1.3 引物设计
利用甲壳动物cox1,nad5和Cytb 3种编码基因的通用引物扩增出 3个片段[14-16]。然后在 GenBank中选取与杂色龙虾线粒体基因组同源性比较高的 3个物种(中国龙虾(NC014339)、日本龙虾(Panulirus japonicus)(NC004251)和锦绣龙虾(Panulirus ornatus)(NC014854)[17-19])的序列进行序列比对, 选取保守性较高的区域, 利用Primer 5.0 软件设计了19对PCR引物(表 1), 覆盖杂色龙虾线粒体基因组全序列, 引物由上海生工生物技术服务有限公司合成。
1.4 PCR扩增
PCR 反应体系: 反应总体积为 25 μL, 其中,10×Buffer 2.5 μL, dNTP2 μL, 上下游引物各 1 μL,TaqDNA聚合酶0.15 μL, DNA模板1 μL, 去离子水18.35 μL。反应程序为: 95℃预变性3 min; 94℃变性20 s, 48~58℃退火50 s, 72℃延伸90 s, 35个循环; 最后72℃延伸10 min。
1.5 扩增产物的纯化回收及测序
取PCR产物2 μL在1%的琼脂糖凝胶中进行电泳, 凝胶成像仪拍照记录。用QIAquick Gel Extraction 50柱式胶回收试剂盒纯化扩增产物, 纯化的步骤按试剂盒说明书进行。取适量纯化产物作为测序反应的模板, 委托上海生工生物技术服务有限公司进行测序。
1.6 序列拼接及分析
所测得序列用Lasergeneversion 7.0(DNASTAR)软件包进行分析, 用Seqman对重叠群进行拼接, 然后用Clustal W软件对拼接后的序列进行人工校对。参考中国龙虾、日本龙虾和锦绣龙虾的线粒体基因组 DNA数据对所获得序列的蛋白编码基因、rRNA和tRNA基因进行识别, 并通过tRNAscan-SE搜索服务器(http: //lowelab.ucsc.edu/t RNAscan-SE/)[20]进行识别tRNA的再验证。利用MEGA5.0[21]分析各种区段的碱基组成特征, 密码子使用频率, 通过 DnaSP 4.10.7[22]分析龙虾属线粒体基因组的编码基因多态位点和基因变异特征。
2 结果与分析
2.1 杂色龙虾线粒体基因组全序列的基本特征
杂色龙虾mtDNA全长为15 700 bp(GenBank登录号为JQ320274), 由13个蛋白质编码基因、22个tRNA基因、2个rRNA基因(16S rRNA和12S rRNA)以及D-loop控制区组成(表2)。其中14个基因位于轻链(L)上, 分别为12S rRNA, 16S rRNA,nad1,nad4L,nad4,nad5,tRNAVal,tRNALeu-CUN,tRNAPro,tRNAHis,tRNAPhe,tRNACys,tRNATyr,tRNAGln, 其余23个基因位于重链(H)上。

表2 杂色龙虾线粒体基因组组成Tab.2 Gene organization of P. versicolor mitochondrial genome
杂色龙虾线粒体基因组存在11处基因重叠, 重叠区长度1~6 bp不等, 基因间隔有10处, 大小在1 ~36 bp,基因间隔最大的在tRNASer与nad1和nad1与tRNALeu之间, 分别是 17 bp和 36 bp; 既没有重叠,也没有间隔的基因共计15处。杂色龙虾线粒体DNA的碱基组成具有AT偏好性, A+T含量为67%, 4种碱基的含量分别为(A=34.6%, T=32.1%, C=20.5%, G=12.8%), 变化趋势与其他甲壳动物基本一致, A含量最高而G含量最低, 在A+T富集区表现更为明显。
线粒体基因排列能够提供重要的系统进化信息,一般而言, 泛甲壳动物的线粒体基因组的基因排列顺序基本相同, 但十足目较为复杂。作者研究的杂色龙虾保持了泛甲壳动物线粒体基因组的原始排列,没有出现重组/随机丢失现象。
2.2 蛋白编码基因
在杂色龙虾线粒体基因组中, 参与蛋白质编码的基因有13个(表3), 核苷酸共11140 bp, 占全序列的70.96%。杂色龙虾的atp6与atp8之间和nad4与nad4L之间的重叠情况一致, 均重叠 7个碱基。除cox1和nad2外, 其余的11个蛋白质编码基因都以标准的ATN作为起始密码子。nad2的起始密码子为GTG,cox1的起始密码子为ACG。除atp6、atp8、cox3、nad1、nad2、nad4L和nad6的终止密码子是TAA外, 其余的终止密码子为不完全的T或TA,在后生动物中这种不完全密码子比较常见。Ojala等[23]的研究表明, 成熟的终止密码子 TAA 可以通过转录后的多腺苷酸化产生。

表3 杂色龙虾粒体基因组13个蛋白编码基因密码子使用情况Tab.3 Average codon frequencies and usage in the 13 mitochondrial protein-coding genes in P. versicolor
2.3 tRNA基因与rRNA基因
杂色龙虾mtDNA序列与其他物种一样, 也包含22个tRNA基因。除tRNALeu和tRNASer有2个, 其他18个均只有1个序列。每个tRNA均能折叠成三叶草状二级结构, 长度为61~75 bp不等。12s rRNA基因位于tRNAVal和 D-loop之间, 长度为 861 bp, 16s rRNA基因位于tRNALeu和tRNAVal之间, 长度为1 460 bp。两个rRNA基因都位于轻链上, 其方向与龙虾属的其他物种完全一致。本研究成功地预测出 18个tRNA基因的二级结构和两个rRNA基因的二级结构。预测到的12s rRNA的二级结构含有3个结构域, 40个茎环结构, 它的3'端存在一个茎环结构; 16s rRNA二级结构都有6个结构域, 53个茎环结构。
在相近种种间或种内的遗传变异分析研究中,选取分子标记片段至关重要。本研究在中国龙虾, 杂色龙虾, 波纹龙虾 3个物种线粒体基因组全序列基础上分析了龙虾属线粒体基因组13个蛋白质编码基因、D-loop及2个核糖体RNA 基因的多态位点分析(表 4)。结果显示: 多态位点比例较大的基因有nad2、nad4、nad5、Cytb、D-loop。
3 讨论与结论
3.1 杂色龙虾线粒体基因组全序列
杂色龙虾线粒体基因组序列全长15 700 bp, 稍大于中国龙虾[18]的15 677 bp, 而稍小于日本龙虾[17]的15 717 bp, 在软甲亚纲的范围内。杂色龙虾线粒体基因组的基因排列次序与其他龙虾类及节肢动物的基本模式相同, 没有KD倒置现象, 即tRNAAsp(D)排在tRNALys(K)基因上游(DK排列)[24]。杂色龙虾线粒体DNA的碱基组成具有很强的 AT偏好性, A+T含量为 67%, 稍高于中国龙虾的 65.6%和日本龙虾的64.5%。4种碱基的含量分别为(A=34.6%, T=32.1%,C=20.5%, G=12.8%), 变化趋势与其他甲壳动物基本一致, A含量最多而G含量最少, 在A+T富集区表现更为明显, 这与线粒体含有大量 ATP有着密切的联系。蛋白质基因nad4和nad4L,atp6、atp8和cox3紧密相连, 没有tRNA基因的间隔, 这也说明存在于蛋白质编码基因间的tRNA二级结构是多顺反子, 在转录过程中不进行蛋白质编码基因间隔序列的形成[25]。
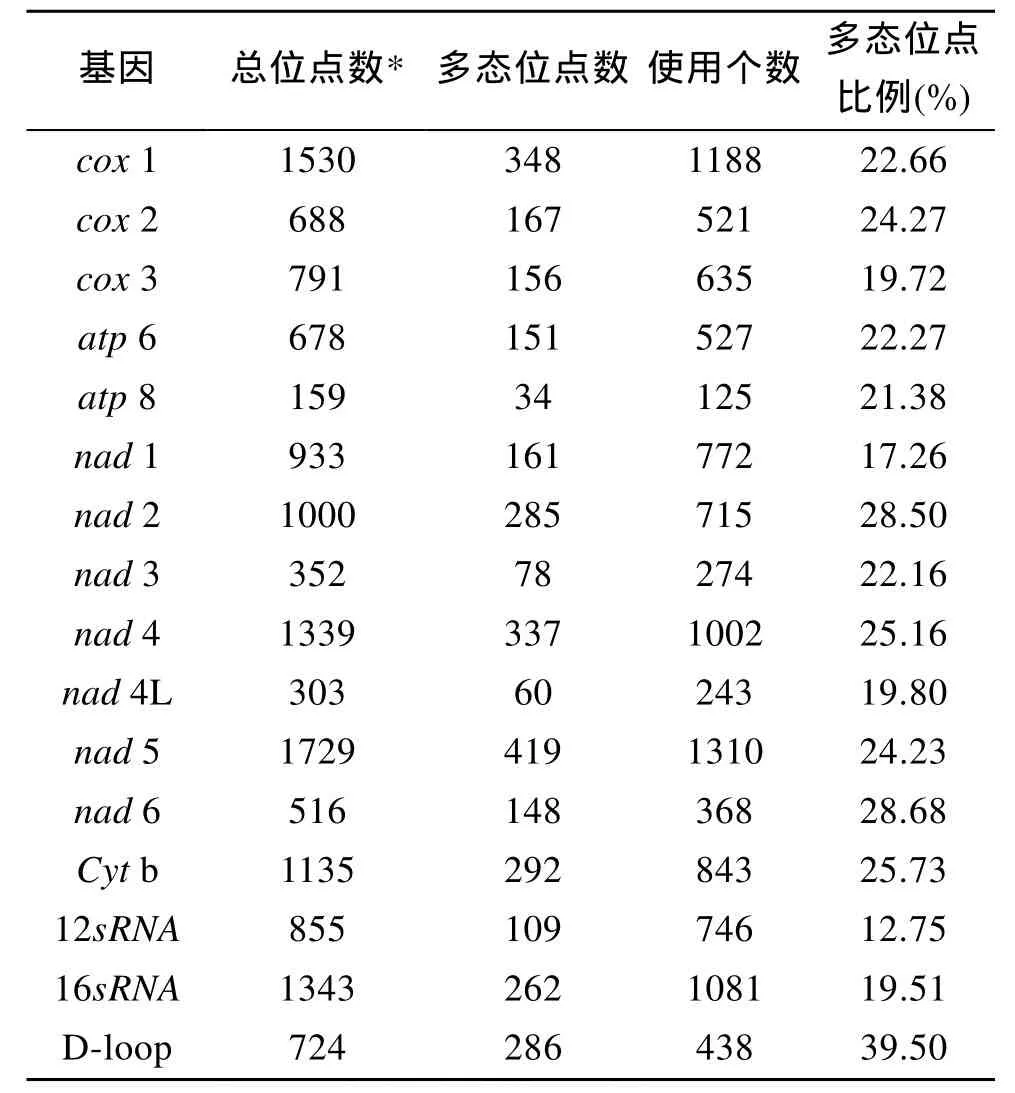
表4 龙虾属线粒体基因组13个蛋白质编码基因、D-loop及2个核糖体RNA基因的多态位点分析Tab.4 Polymorphic loci analysis of 13 protein-coding genes, D-loop and two ribosomes RNA gene in Lobster (Panulirus) mitochondrial genome
3.2 蛋白质编码基因
与日本龙虾及中国龙虾一样, 杂色龙虾的atp6与atp8之间和nad4与nad4L之间均重叠7个碱基。除cox1的起始密码子为ACG、nad2的起始密码子为GTG外, 其余的11个蛋白质编码基因都以标准的ATN作为起始密码子。除atp6、atp8、cox3、nad1、nad2、nad4L和nad6的终止密码子是TAA外, 其余的终止密码子为不完全的T或TA, 在后生动物中这种不完全密码子比较常见。Ojala等[23]的研究表明,成熟的终止密码子 TAA 可以通过转录后的多腺苷酸化产生。
从表 3可以看出, 杂色龙虾组成蛋白质的氨基酸中按数目的多少排列分别为: Ser, Leu, Ile, Phe, Tyr,Pro, Asn, Thr, Lys, Met, Gln, Val, His, Ala, Glu, Arg,Gly, Asp, Trp, Cys。这与已知的龙虾属其他物种的氨基酸排列顺序大致相同, 说明龙虾属各物种的蛋白质编码基因序列之间的替换多为同义替换, 没有影响所编码的氨基酸种类。在杂色龙虾线粒体基因组中, 占主导地位的是非极性氨基酸, 其次是极性不带电的氨基酸。根据生物分子的相似相溶性原理可知, 其线粒体DNA所编码的蛋白质为疏水蛋白质。另外, 从编码同一个氨基酸不同密码子的使用个数和百分比来看, 三联密码子的第三位点有 4种摆动形式, 第三位点为 G的密码子使用个数较少, 这也反映了线粒体蛋白质编码基因在使用密码子上的明显偏好, 即反G偏倚。
3.3 tRNA基因与rRNA基因
杂色龙虾 mtDNA也编码 22个tRNA基因。tRNALeu和tRNASer有 2个序列, 其他 18个均只有 1个序列。长度范围为 61~75 bp。所预测到的二级结构, 仅tRNASer无DHC臂, 由7个核苷酸取代了该位置, 其余的tRNA都能形成典型的三叶草结构, 这是后生动物线粒体基因组的一个普遍特征。杂色龙虾的18个tRNA的二级结构中出现16处错配, 其中14处为 GU错配, 2处 UU错配, 发生在tRNATyr和tRNAGln的氨基酸接受臂和TψC臂。有学者认为线粒体基因组tRNA基因的部分错配可以通过RNA编辑校正, 不会引起氨基酸转运上的障碍[26]。12s rRNA基因位于tRNAVal和D-loop之间, 长度为861bp, 16s rRNA基因位于tRNALeu和tRNAVal之间, 长度为1 460 bp。两个rRNA基因都编码在轻链上, 其方向与龙虾属的其他物种完全一致[17-18]。预测到的杂色龙虾 16s rRNA二级结构, 有6个结构域, 53个茎环结构; 12srRNA的二级结构含有 3个结构域, 40个茎环结构,它的3'端存在一个茎环结构, Cannone等[27]认为12s rRNA和类似12srRNA的二级结构都存在该茎环结构。杂色龙虾的rRNA二级结构与其他脊椎动物的基本相同[28], 这说明rRNA基因进化非常慢, 在相当长的进化过程中, 其分子的排列变化不大, 功能几乎保持恒定。
3.4 A+T富集区
目前对A+T富集区域的关注主要集中于所包含的与复制相关的调控信息。由于其在进化过程中选择压力相对较小, 较线粒体其他区域具有更高的多态性, 不同种类动物线粒体DNA大小的差异主要是由该区域的变化造成的[29]。杂色龙虾控制区与其他后生动物的相似, 主要包括5个结构: 轻链复制起点(origin of minoritystrand replication)、poly T(poly-thymidine stretch)结构、高度变异区、类似微卫星的重复序列以及靠近tRNAIle上游的一段 poly T结构[30-31],这段Poly(T)结构, 与复制起点的识别有关[32]。
3.5 基因多态位点分析
通常cox1、Cytb被认为适用于近缘物种的区分和鉴定, 在群体遗传、分类学、系统进化、动物保护生物学以及古生物化石等研究方面得到了广泛的应用[33-35]。本研究中, 通过对多态位点的分析得知:cox1的多态位点比例较低, 不适宜用于龙虾类分子进化研究。nad2、D-loop基因的多态位点比例较高, 尤其是D-loop, 高达39.50%, 且基因片段较长, 而nad5基因拥有的多态位点总数最多(>400), 因此, 他们比较适合作为分子标记用于分析龙虾类生物进化过程中的谱系发生和迁移流动以及群体间线粒体基因组进化全序列比较分析、系统发育进化、种内多态性研究、遗传分化等。
[1] 李冬玲. 线粒体病的母系遗传[J]. 潍坊学院学报,2006, 6(4): 85-87.
[2] Moriyama E N, Powell J R. Synonymous substitution rates in Drosophila: mitochondrial versus nuclear genes[J]. Journal of Molecular Evolution, 1997, 45(4):378-391.
[3] Caccone A, Gentile G, Burns C E, et al. Extreme difference in rate of mitochondrial and nuclear DNA evolution in a large ectothermGalapagos tortoises[J].Molecular Phylogenetics and Evolution, 2004, 31(2):794-798.
[4] Brown W M, George M, Wilson A C. Rapid evolution of animal mitochondrial DNA[J]. Proceedings of the National Academy of Sciences of the United States of America, 1979, 76(4): 1967-1971.
[5] 刘丽, 李晓娜, 陈育盛, 等. 基于线粒体基因的石珊瑚分子系统学研究[J]. 海洋与湖沼, 2012, 43(4):814-820.
[6] 刘丽, 李文娟, 刘楚吾. 广东徐闻地区滨珊瑚遗传多样性和系统发生关系[J]. 海洋与湖沼, 2013, 44(2):371-376.
[7] 廖顺尧, 鲁成. 动物线粒体基因组研究进展[J]. 生物化学与生物物理进展, 2000, 27(5): 508-512.
[8] Wolstenholme D R. Animal mitochondrial DNA: structure and evolution[J]. International Review of Cytology,1992, 141: 173-216.
[9] Beagley C T, Okimoto R, Wolstenholme D R. The mitochondrial genome of the sea anemoneMetridium senile(Cnidaria): introns, a paucity oftRNAgenes, and a near-standard genetic code[J]. Genetics, 1998, 148(3):1091-1108.
[10] 牛屹东, 李明, 魏辅文, 等. 线粒体 DNA 用作分子标记的可靠性和研究前景[J]. 遗传, 2001, 23(6):593-598.
[11] 刘楚吾, 徐田军, 刘丽, 等. 笛鲷属(Lutjanus)鱼类线粒体16s rRNA基因序列比较及系统学分析[J]. 海洋与湖沼, 2009, 40(5): 563-571.
[12] Liu X, Guo Y, Wang Z, et al. The complete mitochondrial genome sequence ofTrichiurus nanhaiensis(Perciformes: Trichiuridae) [J]. Mitochondrial DNA,2013, 24(5): 516-517.
[13] 卢圣栋. 现代分子生物学实验技术(第2版)[M]. 北京:中国协和医科大学出版社, 1999: 61-66.
[14] Folmer O, Black M, Hoeh W, et al. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates [J].Molecular Marine Biology and Biotechnology, 1994, 3:294-299.
[15] Lavrov D V, Brown W M, Boore J L, et al. Phylogenetic position of the Pentastomida and (pan) crustacean relationships[J]. Proceedings of the Royal Society B, 2004, 271(1538): 537-544.
[16] Boore J L, Brown W M. Mitochondrial genomes ofGalathealinum,Helobdella, andPlatynereis: sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa[J]. Molecular Biology and Evolution,2000, 17(1): 87-106.
[17] Yamauchi M, Masaki U M, Mutsumi N. Complete mitochondrial DNA sequence of the Japanese spiny lobster,Panulirus japonicus(Crustacea: Decapoda) [J].Gene, 2002, 295 (1): 89-96.
[18] Liu Y, Cui Z. Complete mitochondrial genome of the Chinese spiny lobsterPanulirus stimpsoni(Crustacea:Decapoda): genome characterization and phylogenetic considerations [J]. Molecular Biology Reports, 2011,38(1): 403-410.
[19] Liang H F. Complete mitochondrial genome of the ornate rock lobsterPanulirus ornatus(Crustacea:Decapoda) [J]. African Journal of Biotechnology, 2012,11(80): 14519-14528.
[20] Newman S J, Cappo M, Williams D M. Age, growth and mortality of the stripey,Lutjanus carponotatus(Richardson) and the brown-stripe snapper,L. vitta(Quoy and Gaimard) from the central Great Barrier Reef, Australia [J]. Fishery Research, 2000, 48(3):263-275.
[21] Koichiro T, Daniel P, Nicholas P, et al. MEGA5:Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods[J]. Molecular Biology and Evolution, 2011, 28 (10): 2731-2739.
[22] Rozas J, Sanche E L. DNA polymorphism analyses by the coalescent and other methods [J]. Biomedical Informatics, 2003, 19(18): 2496-2497.
[23] Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria [J].Nature, 1981, 290(5806): 470-474.
[24] Ballard J W O, Dean M D. The mitochondrial genome:mutation, selection and recombination [J]. Current Opinion in Genetics and Development, 2001, 11(6):667-672.
[25] Kurabayashi A, Ueshima R. Complete sequence of the mitochondrial DNA of the primitive Opisthobranch gastropodPupa strigosa: systematic implication of the genome organization [J]. Molecular Biology and Evolution, 2000, 17(2): 266-277.
[26] Yokobori S, Paabo S. Transfer RNA editing in land snail mitochondria [J]. Proceedings of the National Academy of Sciences of the United States of America,1995, 92(22): 10432-10435.
[27] Cannone J J, Subramanian S, Schnare M N, et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal intron and other RNAs [J]. BMC Bioinfor-matics, 2002, 3(1): 15.
[28] Hixson J E, Brown W M. A comparison of the small ribosomal RNA genes from the mitochondrial DNA of the great apes and humans: sequence, structure evolution and phylogenetic implications[J]. Molecular Biology and Evolution, 1986, 3(1): 1-18.
[29] Zhang D X, Hewitt G M. Insect mitochondrial control region: a review of its structure, evolution and usefulness in evolutionary studies[J]. Biochemical Systematics and Ecology, 1997, 25(2): 99-120.
[30] Lee W J, Conroy J. Structure and evolution of teleost mitochondrial control regions[J]. Molecular Iology and Evolution, 1995, 41(1): 54-66.
[31] Giles R E, Blane H. Maternal inheritance of human mitochondrial DNA[J]. Proceedings of the National Academy of Sciences of the United States of America,1980, 77(11): 6715-6719.
[32] Ye W, Dang J P, Xie L D, et al. Complete mitochondrial genome ofteleogryllus emma(Orthoptera:Gryllidae) with a new gene order in Orthoptera[J].Zoological Research, 2008, 29(3): 236-244.
[33] Ferran P. Phylogenetic relationships between spiny,slipper and coral lobsters (Crustacea, Decapoda,Achelata) [J]. Molecular Phylogenetics and Evolution,2009, 50(1): 152-162.
[34] George R W, Main A R. The evolution of spiny lobsters(Palinuridae): a study of evolution in the marine environment[J]. Society for the Study of Evolution,1967, 21(4): 803-820.
[35] George R W. Tethys origin and subsequent radiation of the spiny lobsters (Palinuridae) [J]. Crustaceana, 2006,79(6): 397-422.
猜你喜欢
广西植物(2022年8期)2022-09-07
作文大王·低年级(2022年2期)2022-02-28
小火炬·阅读作文(2019年4期)2019-08-06
发明与创新·中学生(2019年6期)2019-06-26
生物学教学(2018年2期)2018-08-07
安徽农业科学(2018年1期)2018-05-14
电脑知识与技术(2017年10期)2017-06-05
电子技术与软件工程(2017年8期)2017-05-10
科技视界(2016年27期)2017-03-14
数学大王·低年级(2015年10期)2015-10-21
