
三种席夫碱-Ni(II)配合物的电子结构和吸收光谱的理论计算
2014-10-11 06:20:10李洁琼赵清岚
化学研究 2014年5期
李洁琼,赵清岚
(河南大学 化学化工学院,河南 开封475004)
席夫碱含有氧(硫)和氮原子,可以与多种过渡金属配位,在配位化学中占有重要的地位,是配位化学的热点研究内容之一[1-3].例如席夫碱可以与Cu(II)、Co(II)、Zn(II)及 Ni(II)等金属形成相应的金属配合物,这些金属配合物具有极好的生物活性,如抗菌、抗真菌、抗癌和抗病毒,还具有广泛的工业用途[4].因此基于席夫碱和其金属配合物的研究受到了来自材料科学、催化和化学反应等多领域科学家的关注.通过加入不同的取代基可以改变席夫碱衍生物和其金属配合物的性质[5],多年来大量的席夫碱衍生物和其金属配合物被相继合成出来.
ASADI等人[6]合成了一系列席夫碱衍生物的镍配合物A、B和C(结构见图1),并对其性质进行了表征,然而他们并没有给出这些化合物的几何结构和详细的光谱信息.研究金属离子和配体之间的配位方式并得到详细的电子光谱,能够为合成新的金属配合物提供可靠的依据.本文作者采用密度泛函理论优化了这三种配合物的几何构型,接着在TD-B3LYP水平下计算了这些配合物的电子光谱,并对其光谱特性进行了归属.
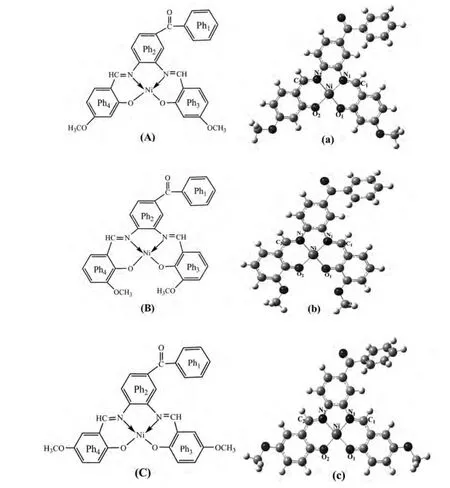
图1 配合物A、B和C的几何结构Fig.1 The schematic and optimized geometric structures of complexes A,Band C
1 计算方法
采用B3LYP[7-8]方法优化了三种配合物的几何构型,对非金属元素使用的是6-31+G(d)基组[9-10],对过渡金属Ni使用LANL2DZ赝势基组[11-13].在相同的水平下进行了频率计算以验证所优化结构的性质,所得的频率全部为实频,表明优化的构型是稳定构型.基于优化的几何构型,采用含时密度泛函(TDB3LYP)[14-15]理论在相同的基组下结合极化连续介质模型(PCM)[16-17]计算了配合物在 N,N-二甲基甲酰胺(DMF)溶液中的电子结构和电子吸收光谱.所有的计算都是在Gaussion 09程序包[18]下完成.
2 结果和讨论
2.1 几何构型
在B3LYP/6-31+G(d)-LANL2DZ水平下优化的主要键长、键角和二面角参数列于表1.由表1可知,A、B和C这三个配合物的结构相似,具有相似的Ni-N1、Ni-N2、Ni-O1和Ni-O2键长和键角,因此在配体的不同位置引入甲氧基对几何构型的影响很小.另外在这三个配合物中,N1-Ni-O2、N2-Ni-O1和N1-N2-O1-O2都接近于180°,所以中心原子Ni与周围的N和O原子形成一个四配位的平面构型,这与实验结果[6]相吻合.

表1 在B3LYP/6-31+G(d)-LANL2DZ水平下优化的配合物A、B和C的几何参数Table 1 The optimized geometry parameters of complexesA,Band Cat B3LYP/6-31+G(d)-LANL2DZ level
2.2 前线分子轨道
前线分子轨道(FMOs)包括分子中能量最高的占有轨道HOMO(H)和能量最低的空轨道LUMO(L),其电子云的分布对分子的电子结构和光谱性质起着决定性作用.图2绘出了三种配合物的部分分子轨道能级,并给出了几个在吸收光谱中涉及的重要分子轨道的轮廓图.各配合物的轨道电子云分布数据列于表2中.从表2可看出配合物A、B和C的HOMO轨道电子云分布特点大致相同,只是各基团的成分略有差别.相对于配合物B和C来说,配合物A的H-1轨道上Ph2贡献显著增多,而H-2轨道上Ph2贡献可以忽略不计.对于L+2轨道来说,金属Ni的成分按照配合物A→C→B的顺序依次增大.另外,对于三个配合物来说,电子云几乎没有分布在甲氧基上.对于H-L能级差来说,席夫碱上甲氧基取代位置对HOMO轨道的影响大于对LUMO轨道的影响.HOMO轨道的能量按照A(-5.88eV)→B(-5.58eV)→C(-5.44eV)的顺序依次增加,即对应甲氧基分别处在间位、邻位和对位.这是因为甲氧基处于间位时,其吸电子诱导效应大于推电子共轭效应,净结果表现出吸电子性,它的加入使席夫碱配体成为一个更强的电子受体从而降低了配合物A的HOMO轨道能量,而甲氧基处于对位时,其推电子共轭效应大于吸电子诱导效应,净结果表现出推电子性,它的加入使席夫碱配体的电子云密度减小从而提高了配合物C的HOMO轨道能量.甲氧基取代位置的不同对LUMO轨道能量的影响很小,因此H-L能级差按照A→B→C的顺序降低.

表2 在B3LYP/6-31+G(d)-LANL2DZ 水平上,配合物A、B、C 在DMF 中其基态分子轨道电子云分布Table 2 Molecular orbital compositions in the ground state of complexes A, B and C at B3LYP/6-31+G(d)-LANL2DZ level in DMF
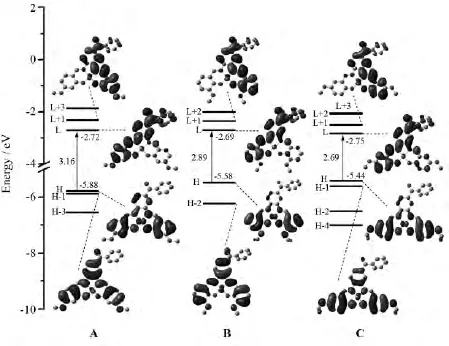
图2 配合物A、B和C的前线分子轨道能量及其能级差Fig.2 Presentation of energy levels,energy gaps,and frontier molecular orbitals for complexes A,Band C
2.3 电子吸收光谱
在DMF中,采用 TD-B3LYP/6-31+G(d)-LANL2DZ//B3LYP/6-31+G(d)-LANL2DZ方法得到了三种配合物的电子吸收光谱,绘于图3.配合物A、B和C能量最低的跃迁按照A→B→C顺序呈现出红移的趋势,这与他们H-L能级差的变化规律是一致的.A、B和C能量最低的跃迁主要源于H-1→L,H→L和H-1→L的跃迁.在340~450nm的区间内,配合物A存在三个强度相近的吸收峰,配合物B有两个强吸收峰,而配合物C有两个强吸收峰和一个中强吸收峰.计算得到的吸收峰值与实验值符合的较好.另外理论计算还确认了实验上没有测到的吸收峰,例如配合物B和C分别在350(振子强度f=0.458 8)和338nm(f=0.390 4)拟合到了强的理论峰,但实验上对此却没有进行报道.得到通过实验手段难于测定的吸收峰或者分离通过实验方法难于确认的肩峰是理论计算的优点之一.另外,理论计算还可以给出详细的光谱跃迁机理,配合物A在389nm处的吸收峰主要源自H-1→L+1的跃迁,由于H-1轨道的电子云主要分布在Ph2,Ph3和Ph4基团上,而L+1轨道的电子云主要分布在Ph2,Ph3和C1基团上,所以该跃迁为(π)→(p,π*)跃迁,具有配体内的电荷转移(ILCT)性质.其他跃迁的详细信息都列于表3中,不再逐一叙述.

图3 在TD-B3LYP(PCM)水平下,配合物A、B和C在DMF溶剂中的吸收光谱曲线Fig.3 Simulated absorption spectra in DMF medium for complexes A,Band Cfrom the TD-B3LYP(PCM)calculation

表3 在TD-B3LYP水平下,配合物A、B和C在DMF溶剂中的吸收光谱数据以及相应的实验值Table 3 Calculated absorptions of complexes A,B,and Cin DMF medium at TD-B3LYP level together with experimental values
3 结论
用B3LYP/6-31+G(d)-LANL2DZ方法优化得到了配合物A、B和C基态的几何构型.研究结果表明,配体上甲氧基取代位置的不同对几何结构影响不大.而间位甲氧基加入增大了配合物A HOMO与LUMO间的能级差,对位甲氧基的加入降低了配合物C HOMO与LUMO间的能级差,从而导致三种配合物能量最低的跃迁按照A→B→C的顺序呈现红移.理论计算的吸收光谱与实验值符合的很好,并确认了实验上没有测到的吸收峰.
[1]AKBARI A,ALINIA Z.Comparative analysis of the Ni(II)complex of the N,N′-bis-(4-Hydroxysalicylidene)-1,2-diaminoethane:combined experimental and theoretical study(DFT/PW91)[J].Comput Res,2013,1(2):19-26.
[2]KOCYIGIT O,KURSUNLU A N,GULER E.Complexation properties and synthesis of a novel Schiff base with triphenylene nucleus[J].J Hazard Mater,2010,183(1):334-340.
[3]BHOWMIK P,DREW M G B,CHATTOPADHYAY S.Synthesis and characterization of nickel(II)and copper(II)complexes with tetradentate Schiff base ligands[J].Inorg Chim Acta,2011,366(1):62-67.
[4]SHAKIR M,KHANAM S,AZAM M,et al.Template synthesis and spectroscopic characterization of 16-membered[N4]Schiff-base macrocyclic complexes of Co(II),Ni(II),Cu(II),and Zn(II):in vitro DNA-binding studies[J].J Coord Chem,2011,64(18):3158-3168.
[5]ESMAIElZADEH S,AZIMIAN L,SHEKOOHI K,et al.Nickel(II)complexes of the unsymmetrical tetradentate Schiff base of methyl-2-(N-20-aminoethane),(1-methyl-20-aminoethane),(3-aminopropylamino)cyclopentenedithiocarboxylate:Synthesis,characterization,thermodynamic and computational studies[J].Inorg Chim Acta,2013,405:155-162.
[6]ASADI M,SEPEHRPOUR H,MOHAMMADI K.Tetradentate Schiff base ligands of 3,4-diaminobenzophenone:Synthesis,characterization and thermodynamics of complex formation with Ni(II),Cu(II)and Zn(II)metal ions[J].J Serb Chem Soc,2011,76(1):63-74.
[7]BECKE A D.Density-functional thermochemistry.III.The role of exact exchange[J].J Chem Phys,1993,98(7):5648-5652.
[8]LEE C,YANG W,PARR R G.Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density[J].Phys Rev B,1988,37:785-789.
[9]LV P C,LI H Q,SUN J,et al.Synthesis and biological evaluation of pyrazole derivatives containing thiourea skeleton as anticancer agents[J].Med Chem,2010,18(13):4606-4614.
[10]PAVAN F R,MAIA P I S,LEITE S R A,et al.Thiosemicarbazones,semicarbazones,dithiocarbazates and hydrazide/hydrazones:anti-Mycobacterium tuberculosis activity and cytotoxicity[J].Eur J Med Chem,2010,45:1898-1905.
[11]WADT W R,HAY P J.Ab initio effective core potentials for molecular calculations.Potentials for main group elements Na to Bi[J].J Chem Phys,1985,82(1):284-298.
[12]HAY P J,WADT W R.Ab initio effective core potentials for molecular calculations.Potentials for K to Au including the outermost core orbitals[J].J Chem Phys,1985,82(1):299-310.
[13]HAY P J,WADT W R.Ab initio effective core potentials for molecular calculations.Potentials for the transition metal atoms Sc to Hg[J].J Chem Phys,1985,82(1):270-283.
[14]BAUERNSCHMITT R,AHLRICHS R.Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory[J].Chem Phys Lett,1996,256(4):454-464.
[15]STRATMANN R E,SCUSERIA G E,FRISCH M J.An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules[J].J Chem Phys,1998,109(19):8218-8224.
[16]MIERTUS S,SCROCCO E,TOMASI J.Electrostatic interaction of a solute with a continuum.A direct utilizaion of ab initio molecular potentials for the prevision of solvent effects[J].Chem Phys,1981,55(1):117-129.
[17]TOMASI J,PERSICO M.Molecular interactions in solution:an overview of methods based on continuous distributions of the solvent[J].Chem Rev,1994,94(7):2027-2094.
[18]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09[CP].Revision A.02,Gaussian,Inc.,Wallingford CT,2009.
猜你喜欢
高中数理化(2023年6期)2023-08-26 13:28:24
读写算(2020年32期)2020-12-17 06:38:28
西部探矿工程(2018年9期)2018-09-11 10:15:44
丝路视野(2018年1期)2018-05-14 09:06:03
中学课程辅导·教师教育(上、下)(2017年12期)2017-07-01 02:26:00
高校招生(2017年5期)2017-06-30 10:49:44
电子制作(2017年1期)2017-05-17 03:54:38
电源技术(2015年12期)2015-08-21 08:58:30
邵阳学院学报(自然科学版)(2015年2期)2015-06-05 12:22:39
无机化学学报(2014年7期)2014-02-28 17:32:28

- 化学研究的其它文章
- 共混硫化丁腈橡胶-氯丁橡胶的物理性能
- 不同消解方法分析污泥中重金属含量的比较
- Synthesis and characterization of a dysprosium-organic framework incorporating 2-hydroxyl-6-methyl-isonicotinic acid and oxalate mixed ligands
- Crystal structure and proton-conductivity of a complex based on phosphomolybdic acid and 2-(2-hydroxybenzene)benzimidazole
- 利用高浓度秸秆废弃物发酵产氢
- 纳米材料在蛋白质分离中的应用