
重甲基SILAC技术分析雄激素依赖性和非依赖性前列腺癌细胞组蛋白H3甲基化的差异*
2014-08-09 01:25王启林李瑞乾金从国张国颖雷永虹
中国病理生理杂志 2014年9期
王启林, 李瑞乾, 金从国, 赵 斌, 张国颖, 白 宇, 雷永虹△
(1昆明医科大学第三附属医院泌尿外科, 2云南省肿瘤研究所,云南 昆明 650118)
前列腺癌在演变过程中可以发生雄激素依赖性向非依赖性的转变,DNA甲基化和组蛋白修饰等表观遗传机制在其中起着重要作用[1]。研究发现组蛋白H3的甲基化和乙酰化与前列腺癌的发展具有明显的相关性[2]。由于组蛋白甲基化是多位点(赖氨酸/K和精氨酸/R)、多形式(一甲基化、二甲基化和三甲基化)的,以往关于前列腺癌中组蛋白H3的甲基化主要是针对某些既定位点进行分析,是否还存在未知的修饰位点和修饰模式以及它们在雄激素依赖性和非依赖性前列腺癌中是否有所不同,这些问题都缺乏全面了解。因此,我们采取可以整体研究蛋白质修饰谱的蛋白质组学策略对此进行分析。通过重甲基细胞培养条件下氨基酸稳定同位素标记技术(stable isotope labeling with amino acids in cell culture,SILAC)[3]结合生物质谱分析雄激素依赖性前列腺癌细胞LNCaP和非依赖性前列腺癌细胞DU145组蛋白H3的甲基化修饰谱,寻找差异修饰位点和模式,探讨前列腺癌从激素依赖性发展为非激素依赖性的表观遗传机制。
材 料 和 方 法
1 主要试剂和仪器
雄激素依赖性前列腺癌细胞LNCaP和非依赖性前列腺癌细胞DU145(中国科学院上海细胞库);有、无甲硫氨酸的RPMI-1640培养基、透析胎牛血清和细胞培养用胰酶(Invitrogen);胎牛血清(Biochrom AG);L-methionine-(methyl-13C, D3) ([13CD3]-甲硫氨酸)、DTT和iodoacetamide(Sigma);蛋白质组用胰酶(Promega);蛋白酶抑制剂(Roche);TransZol Up(中国全式金生物公司);反转录和荧光定量检测试剂(TaKaRa);BCA蛋白浓度测定试剂盒和Ⅰ抗稀释液(中国碧云天公司);兔抗Histone H3,Mono-、Di-、Trimethyl Histone H3(Lys36)和HRP-山羊抗兔IgG(Cell Signaling);Amersham ECL Plus Western Blotting检测试剂(GE)。酶标仪(BioTek);蛋白电泳和转移系统(Bio-Rad);ImageQuantTMLAS 4000(GE);NanoDrop ND-1000紫外分光光度计(Thermo);7500荧光定量PCR仪(Applied Biosystems)。
2 主要方法
2.1蛋白的重甲基SILAC标记 细胞在未标记时的传代培养用10%胎牛血清和含甲硫氨酸的RPMI-1640培养基,培养条件为37 ℃、5% CO2、100%湿度。进行重甲基化标记时改用10%透析胎牛血清和不含甲硫氨酸的RPMI-1640培养基,添加25 mg/L [13CD3]-甲硫氨酸,连续培养6代以上[3]。该技术策略见图1。
2.2组蛋白提取 用酸提取法[4],于4 ℃操作。2种标记后的细胞分别收集5×107个,用PBS洗涤后,加5 mL低渗裂解液重悬,并转移到1.5 mL离心管中,每管1×107个细胞。低渗裂解液pH 8.0,含10 mmol/L Tris-HCl,1 mmol/L KCl,1.5 mmol/L MgCl2和1 mmol/L DTT,使用时添加蛋白酶抑制剂。将离心管放置于旋转仪上,旋转60 min,使细胞在低渗溶液中发生机械剪碎。10 000×g离心10 min沉淀细胞核,用低渗裂解液洗涤1次,每管细胞核沉淀重悬于500 μL 0.2 mol/L H2SO4溶液,旋转过夜。16 000×g离心10 min收集含有组蛋白的上清液,添加33% TCA沉淀组蛋白,在冰上放置30 min。16 000×g离心10 min,去除上清,用冷丙酮洗涤2次(16 000×g离心5 min),待丙酮挥发后,加适量2% SDS溶液溶解组蛋白,用BCA法测定蛋白浓度,-80 ℃保存备用。
2.3组蛋白H3的凝胶分离及酶解 每种细胞的组蛋白样品各取100 μg进行SDS-PAGE,分离胶为12%,根据分子量标准剪切相应的组蛋白H3条带(15.4 kD)。将收集的条带切成1 mm×1 mm×1 mm的小颗粒,ddH2O洗涤3次;用pH 8.9 50%乙腈/50 mmol/L碳酸氢铵溶液脱色后,进行还原(10 mmol/L DTT,37 ℃反应0.5~1 h)和烷基化(55 mmol/L IAA/50 mmol/L碳酸氢铵溶液,37 ℃避光反应0.5 h);经过100%乙腈干胶后,加入适量的10.0 mg/L trypsin溶液(用20 mmol/L碳酸氢铵溶液稀释),37 ℃酶解12 h。用50%乙腈/5% TFA提取肽段3次,分别合并提取液,冻干,-80 ℃保存备用。
2.4质谱鉴定 质谱鉴定由中科新生命生物科技有限公司完成。(1)样品处理:各酶解肽段溶解为200 μL进行C18脱盐,洗脱后肽段离心干燥后溶解于30 μL 0.1% 甲酸水溶液中。(2)毛细管高效液相色谱:取10 μL样本进行纳升液相-串联质谱(nano-LC-MS/MS)分析。流动相A为2%乙腈/0.1%甲酸/98%水,B为98%乙腈/0.1%甲酸/2%水。样品通过100% A相在2 μL/min流速下完成上样,富集于ChromXP C18预柱上(3 μm, C18-CL, 120 Å, 360 μm×0.5 mm),并在300 nL/min的流速下,通过一个240 min的梯度在Thermo scientific EASY column(75 μm×100 mm 3 μm-C18)上得到分离。使用的梯度为:0~50 min,B相由0线性升至50%;50~58 min,B相由50%线性升至100%;58~60 min、 B相保持为100%。肽段经过液相分离后进入质谱进行检测,质谱系统为AB SCIEX Triple TOF 5600质谱。(3)ESI质谱鉴定:串联质谱检测采用的是信息关联扫描(information-dependent acquisition,IDA)模式。TOF MS 扫描分辨率为30 000(FWHM),质荷比范围设定为m/z 350~1 500,累积时间250 ms;峰高超过120 counts/s,且电荷为+2至+5的30个丰度最大的多肽选择被MS/MS分析,质荷比范围为m/z 100~1 250,每个TOF MS/MS扫描累积时间为100 ms,动态排除时间18 s。MS/MS获取时,开启自动计算碰撞能量(autoCE)功能。(4)ESI质谱数据分析:原始文件(raw file)通过Mascot 2.2软件进行数据库检索。数据库为下载于UniProt的人的所有组蛋白的fasta文件(收录序列55条,下载日期为20130609)。搜库参数设定如下: enzyme为trypsin;missed cleavage设为4;静态修饰设定Carbamidomethy C;动态修饰设定Oxidation Met-0(ΔM=15.99492),Oxidation Met-4(13C,D3,ΔM= 20.01710),Monomethyl(MKR,13C,D3,ΔM=18.03784),Dimethyl(KR,13C,D3,ΔM=36.075668),Trimethyl(K,13C,D3,ΔM=54.11351),Acetyl (K,ΔM= 42.01057)。Peptides tolerance设为20 ppm,MS/MS tolerance 设为0.1 D,significance threshold≤0.05,score≥20。

Figure 1. The heavy methyl SILAC strategy. SILAC means “stable isotope labeling with amino acids in cell culture”. A: using “hea-vy” [13CD3]methionine as methyl donor to transfer the heavy methyl groups to methyl acceptors such as proteins, DNA and RNA; B: the cells were grown separately in “light” or “heavy” medium, containing [12CH3]methionine (Met-0) and [13CD3]methionine (Met-4), respectively. This encodes their proteomes with isotopically distinct forms of methionine and methylated residues. The purified histone H3 was digested with trypsin, then analyzed by LC-MS. The mass of heavy methyl is increased 4 D more than that of the light methyl (native methyl), which makes easy to distinguish trimethylation from acetylation, because the mass difference between acetylation (42.0106 D) and native trimethylation (42.0470 D) is very small (0.0364 D), while that of heavy trimethyl is 54.1135 D.
2.5Western blotting验证2种细胞H3K36的甲基化差异 通过Western blotting检测2种细胞组蛋白样品中H3K36甲基化的状态,验证质谱分析结果。Ⅰ 抗和Ⅱ抗分别稀释1 000倍和2 000倍使用。用Bio-Rad Quantity One软件分析各条带的灰度值,将每种甲基化Histone H3的灰度值除以各自总Histone H3的灰度值计算相对含量,再比较2株细胞间的差异。
2.6荧光定量PCR检测H3K36甲基化酶和去甲基化酶在2种细胞中的表达 在细胞状态良好时,收集细胞提取RNA,通过real-time PCR检测相关酶类的表达量。RNA提取、反转录和real-time PCR均按照试剂说明书进行操作。以β-actin为内参照,采用2-ΔΔCt法计算mRNA的相对表达量,引物见表1。

表1 Real-time PCR引物序列
3 统计学处理
数据以均数±标准差(mean±SD)表示。组间差异采用Excel统计软件进行t检验,以P<0.05为差异有统计学意义。
结 果
1 2种细胞组蛋白H3甲基化的质谱鉴定
质谱鉴定发现2株前列腺癌细胞的组蛋白H3存在5个甲基化位点,甲基化模式分别为H3K14me2、H3R17me1、H3K36me1、H3K36me2、H3K36me3、H3R72me2、H3K79me1和H3K79me2,见图2,全部肽段信息见表2。其中,包含甲基化位点H3K36的肽段“KSAPATGGVKKPHR”鉴定到的频数高于“KSAPSTGGVKKPHR”,两者的区别在于第31位的氨基酸分别为A与S,且在2株细胞间存在明显差异,前者归属于组蛋白H3变异体H31T、H31和H32,主要出现在DU145细胞中,而后者属于组蛋白H3变异体H33,在LNCaP细胞中出现次数稍多;提示2株细胞可能表达不同的组蛋白H3变异体,从而导致甲基化模式的差异。
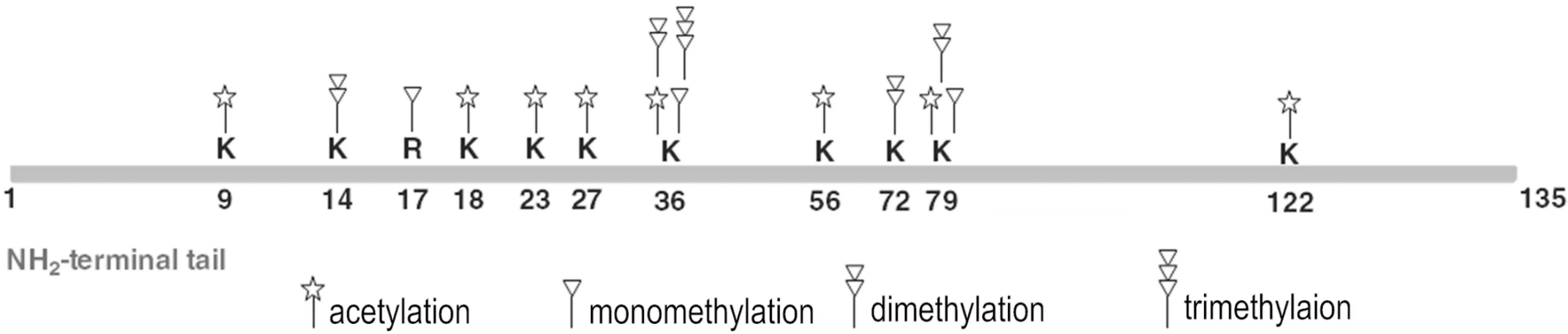
Figure 2. The gobal methylation and acetylation of histone H3 in the 2 cell lines identified by MS. K: lysine; R: arginine.
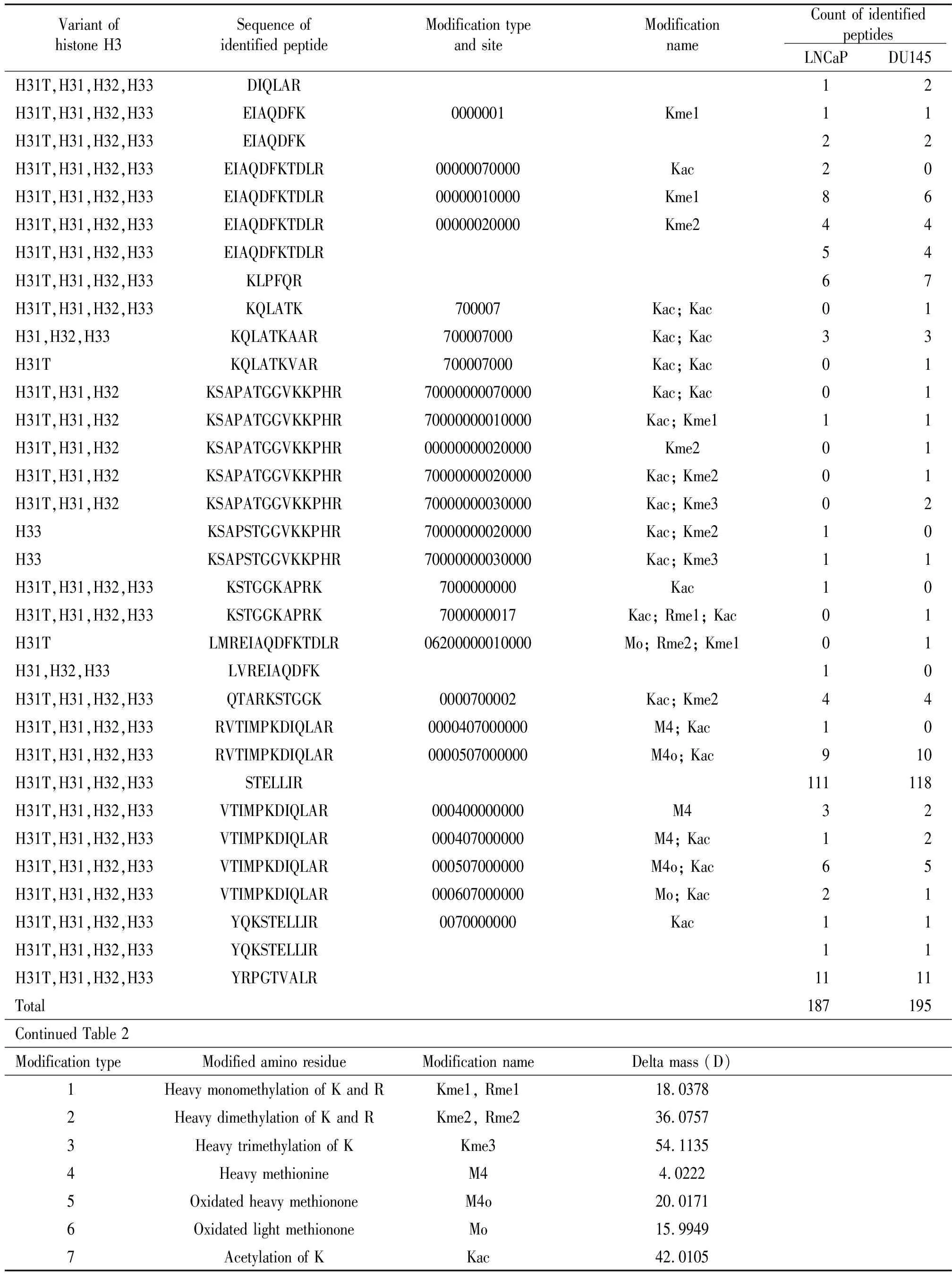
表2 质谱鉴定的肽段在2种细胞间的差异
2 2种细胞组蛋白H3K36甲基化的差异
Western blotting检测结果显示H3K36的一甲基化和二甲基化在2株细胞间没有差异,而DU145细胞H3K36的三甲基化程度显著高于LNCaP细胞,见图3,提示在前列腺癌由激素依赖性发展为非依赖性的过程中,H3K36的三甲基化可能出现特异性地升高,从而影响到相关基因的表达,导致表型变化。
3 H3K36甲基化酶和去甲基化酶在2种细胞中的表达差异
如图4所示,相关的甲基化酶在2种细胞间没有明显差异,但是DU145细胞的去甲基化酶的表达程度比LNCaP细胞有所降低,其中去甲基化酶KDM2B1的表达量降低了12倍之多。这些酶的表达差异可能也是造成2种细胞组蛋白H3甲基化改变的原因之一。
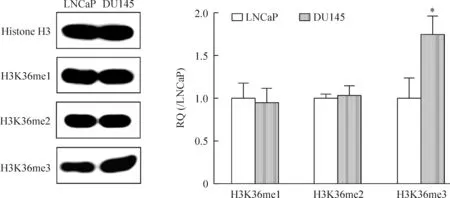
Figure 3. The methylation status of histone H3K36 in the two prostate cancer cell lines LNCaP and DU145. H3K36me1: H3K36 mo-nomethylation; H3K36me2: H3K36 dimethylation; H3K36me3: H3K36 trimethylation. Mean±SD. n=3.*P<0.05 vs LNCaP.
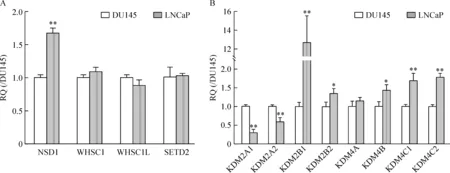
Figure 4. Differential expression of H3K36 methylases (A) and demethylases (B) in the two prostate cancer cell lines LNCaP and DU145 were tested by real-time PCR. NSD1, WHSC1, WHSC1L and SETD2 can mono- and dimethylate H3K36, and SETD2 can also trimethylate H3K36; KDM2A and KDM2B are mono- and didemethylases of H3K36, while KDM4A, KDM4B, and KDM4C are tridemethylases of H3K36. The expression of H3K36 trimethylase SETD2 was not obviously different between the two cell lines, while H3K36 tridemethylases were decreased in DU145 cells compared to LNCaP cells. Mean±SD. n=3.*P<0.05, **P<0.01 vs DU145.
讨 论
前列腺癌在演变过程中可以发生雄激素依赖性向非依赖性的转变,其中涉及大量基因的表达变化,包括激素受体、细胞周期相关基因、DNA损失修复相关基因、信号传导通路相关基因、细胞黏附因子等[1, 5-6]。表观遗传机制是调控基因转录的一类至关重要的事件,包括DNA甲基化和组蛋白修饰等,在生长发育和肿瘤发生中起着重要作用[1-2, 7]。组蛋白可以被进行多种多样的翻译后修饰,例如甲基化、乙酰化、磷酸化、泛素化、SUMO化等,其中甲基化和乙酰化已得到较为深入的研究,相关结果表明其特异的赖氨酸和精氨酸的甲基化和乙酰化对基因表达具有重要调控作用。
组蛋白甲基化是多位点、多形式的,可发生于不同的组蛋白上,也可以发生在不同氨基酸位点上,还可以在同一氨基酸位点上发生一甲基化、二甲基化或三甲基化3种不同的修饰形式。回顾以往的研究,关于前列腺癌中组蛋白的甲基化多为针对某些选定的位点进行分析,那么是否还存在未知的修饰位点和修饰模式以及它们在雄激素依赖性和非依赖性前列腺癌中是否有所不同,这些问题迄今仍缺乏全面的了解。
本研究应用重甲基SILAC技术结合生物质谱全面分析雄激素依赖性前列腺癌细胞LNCaP和非依赖性前列腺癌细胞DU145组蛋白H3的甲基化修饰谱,寻找差异的修饰位点和模式。重甲基是由碳13和氘组成的同位素甲基,比常规甲基(轻甲基)的质量数增加4个道尔顿,在质谱鉴定三甲基化修饰时可以明确区分与常规轻三甲基质量数非常接近的乙酰基(重三甲基54.1135D,轻三甲基42.0470D,乙酰基42.0106D),避免结果误判[8]。通过质谱鉴定,我们发现2株前列腺癌细胞可能表达不同的组蛋白H3变异体(DU145细胞以H31T、H31和H32为主,LNCaP细胞以H33为主),且两者H3K36位点的甲基化也有所不同。Western blotting检测表明H3K36的一甲基化和二甲基化在2株细胞间没有显著差异,而其在DU145细胞中的三甲基化程度显著高于LNCaP细胞。不同的变异体是否会影响H3K36的甲基化还有待研究。
组蛋白甲基化是由组蛋白甲基转移酶将S-腺苷甲硫氨酸的甲基基团转移到组蛋白的赖氨酸或者精氨酸残基上。H3K36的甲基化酶有NSD1(nuclear receptor-binding SET domain-containing protein 1)、WHSC1(Wolf-Hirschhorn syndrome candidate 1)、WHSC1L和 SETD2(SET domain containing 2),这些酶都可以作为H3K36的一、二甲基转移酶,SETD2还可以三甲基化H3K36。去甲基化酶KDM2A[lysine (K)-specific demethylase 2A]和KDM2B可以去除H3K36位点的一、二甲基化,而KDM4A、KDM4B、KDM4C和KDM4D可以去除二、三甲基化[9]。我们应用real-time PCR检测了这些酶在2株前列腺癌细胞中的表达情况,结果显示DU145细胞的H3K36去甲基化酶mRNA表达比LNCaP细胞有所降低,这一差异可能也是造成2种细胞组蛋白H3甲基化改变的原因之一。
组蛋白H3的甲基化对不同基因的表达表现出不一样的调节作用,或激活,或抑制。在LNCaP细胞中,H3K9的甲基化可以抑制雄激素受体靶基因的表达,沉默H3K9的去甲基化酶能够降低PSA、TMPRSS2和NKX 3.1等的表达[10-12];而在CRPC(castration-resistant prostate cancer)细胞模型中,H3K4的甲基化可以促进雄激素受体靶基因的表达,H3K4的一甲基化和二甲基化可以选择性地富集于细胞周期M期基因(UBE2C和CDK1)的雄激素受体增强子上,有助于雄激素受体上调这些基因的表达,促进CRPC生长[13]。临床组织样本的免疫组化研究也提示了组蛋白的甲基化和乙酰化与前列腺癌发生发展的相关性。例如,H3K9Ac、H3K18Ac和H3K4diMe在前列腺癌组织中染色明显[14];在CRPC中,H3K4的3种甲基化都比局限性前列腺癌显著增加[15]。
组蛋白的甲基化在前列腺癌的发生发展中扮演重要角色。综合本实验的研究结果,组蛋白H3变异体和H3K36去甲基化酶的差异表达可能是导致非激素依赖性前列腺癌细胞H3K36三甲基化增加的原因之一,成为前列腺癌从激素依赖性发展为非激素依赖性的一种表观遗传转变,而组蛋白的甲基化能够调控众多基因的表达,可能因此改变了前列腺癌细胞对激素的依赖性,其中更为深入的分子机制还需要进一步探讨。
[参 考 文 献]
[1] Albany C, Alva AS, Aparicio AM, et al. Epigenetics in prostate cancer[J]. Prostate Cancer, 2011, 2011:580318.
[2] Chen Z, Wang LG, Wang QB, et al. Histone modifications and chromatin organization in prostate cancer[J]. Epigenomics, 2010, 2(4):551-560.
[3] Ong SE, Mittler G, Mann M. Identifying and quantifyinginvivomethylation sites by heavy methyl SILAC[J]. Nat Methods, 2004, 1(2):119-126.
[4] Shechter D, Dormann HL, Allis CD, et al. Extraction, purification and analysis of histones[J]. Nat Protoc, 2007, 2(6):1445-1457.
[5] 姜永光, 罗 勇. 前列腺癌雄激素依赖特性转变的生物学机制的研究进展[J]. 现代泌尿外科杂志, 2008, 13(6):411-414.
[6] Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer[J]. Clin Cancer Res, 2006, 12(6): 1665-1671.
[7] Li LC, Carroll PR, Dahiya R. Epigenetic changes in prostate cancer: implication for diagnosis and treatment[J]. J Natl Cancer Inst, 2005, 97(2):103-115.
[8] Li KK, Luo C, Wang DX, et al. Chemical and biochemical approaches in the study of histone methylation and demethylation[J]. Med Res Rev, 2012, 32(4): 815-867.
[9] 杨林森, 张元亮, 黄秋花. 组蛋白H3K36 甲基化与疾病[J]. 生命的化学, 2013, 33(5):543-548.
[10] Yamane K, Toumazou C, Tsukada Y, et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor[J]. Cell, 2006, 125(3):483-495.
[11] Wissmann M, Yin N, Müller JM, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression[J]. Nat Cell Biol, 2007, 9(3):347-353.
[12] Metzger E, Wissmann M, Yin N, et al. LSD1 demethy-lates repressive histone marks to promote androgen-receptor-dependent transcription[J]. Nature, 2005, 437(7057):436-439.
[13] Wang Q, Li W, Zhang Y, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer[J]. Cell, 2009, 138(2):245-256.
[14] Seligson DB, Horvath S, Shi T, et al. Global histone modification patterns predict risk of prostate cancer recurrence[J]. Nature, 2005, 435(30):1262-1266.
[15] Ellinger J, Kahl P, von der Gathen J, et al. Global levels of histone modifications predict prostate cancer recurrence[J]. Prostate, 2010, 70(1):61-69.
猜你喜欢
现代仪器与医疗(2021年6期)2022-01-18
世界科学技术-中医药现代化(2021年12期)2021-04-19
商情(2017年38期)2017-11-28
中国洗涤用品工业(2017年2期)2017-04-16
中国医疗美容(2015年1期)2015-07-12
中国医疗美容(2015年1期)2015-07-12
医学研究杂志(2015年12期)2015-06-10
中国医科大学学报(2015年10期)2015-03-01
郑州大学学报(医学版)(2015年2期)2015-02-27
癌变·畸变·突变(2015年3期)2015-02-27
