
2-(4-三氟甲基苯基)-4-乙基-呋喃-3-酰胺衍生物的设计、合成及其抗肿瘤活性研究
2014-08-06 06:07王甜甜张一凯牛春娟李令振第二军医大学药学院药物化学教研室上海200433
药学实践杂志 2014年2期
张 涛,王甜甜,张一凯,牛春娟,李令振,李 科 (第二军医大学药学院药物化学教研室,上海 200433)
肿瘤特别是恶性肿瘤严重威胁人类健康。恶性肿瘤的预后差、患者痛苦、治疗费用高,病死率超过30%,全球每年约有700万人死于恶性肿瘤,我国每年因肿瘤死亡患者超过160万人。由于环境污染及不良生活习惯,近年来肿瘤尤其是恶性肿瘤的发病率逐年上升,且逐渐呈现出年轻化的趋势。化疗是临床抗肿瘤的主要手段之一。因此,抗肿瘤药物是药物研发的重要方向。
目前上市的抗肿瘤药物主要有以氟尿嘧啶、甲氨蝶呤等为代表的抗代谢药物,以阿霉素、博来霉素等为代表的抗肿瘤抗生素,以及以紫杉醇、长春碱等为代表的抗肿瘤植物药及其他抗肿瘤药物。诸多应用于临床的抗肿瘤药物普遍存在对实体瘤疗效差、毒副作用较大、容易产生多药耐药等缺点。因此,研发更为高效、低毒的新型抗肿瘤药仍是新药研究的热点之一。
呋喃是非常重要的一类杂环有机化合物,广泛存在于各种结构的天然产物中,不仅是抗菌、抗病毒、抗肿瘤的天然产物的骨架和重要组成部分,如Sumiki′s acid,acetyl Sumiki′s acid,perillene, furfuryl thiol等,更是雷尼替丁(ranitidine)和呋喃西林(nitrofurazone)等上市药物不可或缺的药效团[1-4]。
香榧为裸子植物红豆杉科榧属植物榧(Torreyagrandis)的干燥成熟种子,中药以其假种皮入药。香榧中含有多种可在人体中产生药理作用的活性成分,周大铮等[5-6]从香榧假种皮中分离得到11个化合物,其中8个二萜、3个木质素、1个全新2-芳基-4-苄基四氢呋喃木质素化合物(TG-10)。体外活性测试发现,TG-10具有一定的抗HIV病毒作用以及较好的抗肿瘤活性。TG-10结构复杂、合成困难、抗瘤谱较窄。笔者以TG-10为先导化合物,对其进行结构改造和修饰,以对三氟甲基苯甲醛和丙二酸二乙酯为起始原料,经缩合、环合、酰氯化及胺解等4步反应,设计合成了系列2-(4-三氟甲基苯基)-4-乙基-呋喃-3-酰胺目标化合物。采用MTT法,对所设计合成的新化合物进行A549、QGY、HeLa和SW480 4种肿瘤细胞活性测试。结果显示,所设计合成的新型化合物对受试肿瘤株均有较好的抗肿瘤活性,其中5b显示出最优的高效、广谱抗肿瘤活性,值得继续研究。详见图1和图2。

图1 TG-10和目标化合物的结构
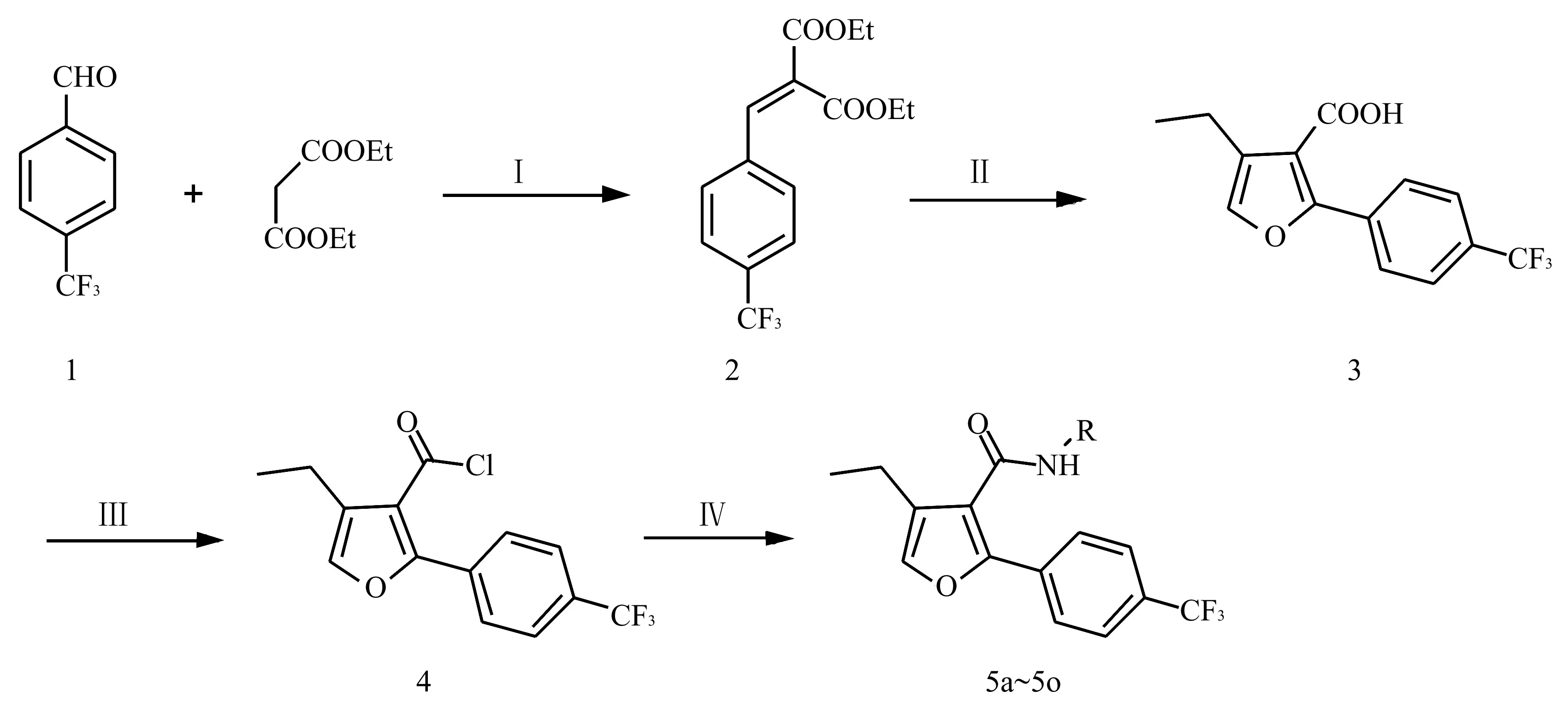
图2 目标化合物的合成路线
1 合成路线
熔点用RY-2型熔点仪测定,温度未经校正;核磁共振用BrukerAC-300P型仪器,300 MHz测定,CDCl3及d6-DMSO为溶剂,TMS为内标;质谱采用Q-TOF Micro YA019质谱仪测定;红外光谱采用Bruker Bector22型红外光谱仪,KBr压片;抗肿瘤活性经酶标仪ELX800测定;柱层析在硅胶H(10~40 μm)上进行;薄层层析在HSGF254型硅胶板(青岛海洋化工厂)上进行;所有试剂均为分析纯。
1.14-三氟甲基苯基次甲基丙二酸二乙酯的合成 三颈瓶中加入对三氟甲基苯甲醛(20 g,0.115 mol)、丙二酸二乙酯(19.32 g,0.121 mol)、冰乙酸(0.34 g,0.006 mol)、哌啶(0.29 g, 0.003 mol)和干燥的甲苯200 ml,搅拌回流反应,过夜。冷凝回流,分出反应过程中产生的水,减压蒸去甲苯,得固体产物,加入50 ml甲醇重结晶。过滤烘干得白色固体34.52 g,收率95.03%,熔点:51.3~51.7 ℃。1H-NMR(300 MHz,CDCl3): δ 8.35(s,1H), 7.74(d,J=8.5 Hz,2H), 7.65(d,J=8.7 Hz,2H), 4.26(m,4H), 1.31(m,6H). ESIMS:m/z(%): 315.07 [M-H]-。
1.22-(4-三氟甲基苯基)-4-乙基-呋喃-3-羧酸的合成 三颈瓶中加入200 ml无水四氢呋喃和1,4-丁炔二醇(8.17 g, 0.095 mol),然后加入NaH(7.59 g, 0.190 mol),室温搅拌至无氢气放出,依次加入4-三氟甲基苯基次甲基丙二酸二乙酯(20 g, 0.063 mol)、碘化亚铜(1.20 g, 0.006 mol),60 ℃反应16 h。反应完全后,加入适量的的水将反应停止,调节pH值至 2~3。减压蒸去四氢呋喃,加入适量的水稀释,二氯甲烷萃取3次,合并二氯甲烷相,减压浓缩,200~300目硅胶柱层析,PE:EA=10:1~2:1洗脱,减压蒸去洗脱液得白色固体 ,收率85.00%,熔点: 159.9~161.0 ℃。1H-NMR(300 MHz,DMSO): δ 12.02(s,1H), 7.95(d,J=8.7 Hz, 2H), 7.75(d,J=8.4 Hz, 2H), 7.63(s, 1H), 2.57(q,J=7.5Hz, 2H), 1.15(J=7.5 Hz, 3H)。13C-NMR(75 MHz,DMSO): δ 165.3, 159.7, 156.0, 138.5, 129.5, 128.6, 126.6, 113.5, 113.0, 55.2, 17.8, 13.9。ESIMS:m/s(%): 283.10 [M-H]-。 IR(cm-1): 2 974, 2 942, 2 882, 2 678, 2 633, 2 583, 2 526, 1 683, 1 620, 1 594, 1 549, 1 507, 1 449, 1 427, 1 384, 1 332, 1 227, 1 170, 1 132, 1 080, 1064, 1 017, 931, 847, 805, 763, 713。
1.32-(4-三氟甲基苯基)-4-乙基-呋喃-3-酰胺的合成 三颈瓶中加入60 ml无水二氯甲烷、2-(4-三氟甲基苯基)-4-乙基-呋喃-3-羧酸(1 g, 0.004 mol)、氯化亚砜(0.62 g, 0.005 mol)回流反应8 h,减压蒸去二氯甲烷,用无水二氯甲烷带走过量的氯化亚砜后制得2-(4-三氟甲基苯基)-4-乙基-呋喃-3-酰氯(4),制得的酰氯用50 ml的无水二氯甲烷溶解后,与不同取代的苄胺、苯胺、苯乙胺及哌嗪衍生物反应,加入无水吡啶作缚酸剂。反应完成后,调节pH值至弱酸性,加入适量的水稀释,二氯甲烷萃取3次,合并二氯甲烷相,减压浓缩后,PE:EA=10:1结晶,得目标化合物5a~5o。
目标化合物的化学结构、熔点、收率、含量及光谱数据见表1。

续表1
2 药理部分
2.1受试药 试验时根据所设剂量组浓度用含10%胎牛血清的Tris-1640培养液配制。
2.2阳性对照药 5-氟尿嘧啶(5-Fu,第二军医大学药学院药物化学教研室提供,4 ℃冰箱保存)。
2.3测试细胞株 人肺癌细胞(A549)、人肝癌细胞(QGY)、人宫颈癌细胞(HeLa)、人结肠癌细胞(SW480)。
2.4试验方法 MTT法检测样品对4种肿瘤细胞的毒性。取对数生长期细胞,用含10%小牛血清的Tris-1640 培养液,制成单细胞悬液1×106/ml,将该悬液加到96孔板中,每孔加入100 μl。于37 ℃培养箱中培养24 h后,吸取上清液,分别加入各浓度的受试药物,设三复孔,继续培养24 h。吸取上清液,加入20 μl MTT溶液(5 μg/ml),继续培养4 h后,吸取上清液,加入100 μl的DMSO,充分溶解后在570 nm处测定吸光值(OD值),并计算其抑制率:抑制率(%) = (对照孔OD值-实验孔OD值)/对照孔OD值×100%,IC50值由SPSS软件计算得出。实验结果见表2。

表2 目标化合物的抗肿瘤活性(IC50,μm)
3 讨论
在合成中间体3时,反应Ⅱ采用一种新颖的、之前未见文献报道的[3+2]domino反应,可通过控制NaOH的量(1.2eq)、反应温度(60 ℃)和延长反应时
间,高选择性、高收率地得到中间体3,从而避免四氢呋喃并[3,4-c]吡喃酮和2,5-二氢呋喃衍生物的生成[7]。
通过对所合成的15个化合物进行抗肿瘤活性测试发现,对于人肺癌细胞A549,多数化合物显示出较好的肿瘤抑制作用。取代苯乙胺形成的酰胺衍生物的活性好于取代苄胺形成的酰胺衍生物,苯胺形成的酰胺衍生物活性最差,哌嗪及4-取代苯基哌嗪形成的衍生物对该肿瘤细胞的抑制作用完全丧失。芳环上对位取代形成的酰胺衍生物的活性优于邻位取代形成的衍生物。化合物5b、5j显示出较好的抑制人肺癌细胞A549的活性,IC50值分别为0.34、3.7 μm,分别达到了阳性对照药5-Fu的16.47倍和1.51倍;对于人肝癌细胞QGY,哌嗪及取代哌嗪形成的酰胺衍生物5m、5n、5o显示了中等的抗肿瘤活性,仅有少量的取代苯胺、取代苄胺形成的酰胺衍生物显示了一定的抗肿瘤活性,多数取代苯胺、取代苄胺和取代苯乙胺对该细胞的活性丧失;对于人宫颈癌细胞(HeLa细胞),取代苯乙胺形成的酰胺衍生物的活性要好于取代苄胺形成的酰胺衍生物的活性,更好于苯胺形成的酰胺衍生物的活性,而哌嗪及4-取代哌嗪形成的酰胺衍生物的活性丧失。其中,化合物5b、5e、5j、5h的 IC50值分别为1.9、2.3、1.3、14.8 μm,达阳性对照药5-Fu的8.47、7.00、12.38和1.09倍; 对于人结肠癌细胞SW480,仅有少量的取代苄胺、取代苯乙胺、取代苯胺形成的酰胺衍生物显示了一定的抗肿瘤活性。值得注意的是,化合物5b、5e、5j显示出高效、广谱的抗肿瘤活性,远远超过阳性对照药5-Fu,值得深入研究。
【参考文献】
[1] Wright DL. Furan as a versatile synthon[J]. Chem Innov,2001, 31(10): 17-21.
[2] Min BS,Yun BS,Lee HK,etal. Two novel furan derivatives fromPhellinuslinteuswith anti-complement activity[J]. Bioorg Med Chem Lett,2006, 16: 3255-3257.
[3] Hough LB,Menge WMPB,Vande Stolpe AC,etal. Antinociceptive activity of furan-containing congeners of improgan and ranitidine[J]. Bioorg Med Chem Lett,2007, 17: 5715-5719.
[4] Zanatta N,Alves SH,Coelho HS,etal. Synthesis, antimicrobial activity, and QSAR studies of furan-3-carboxamides[J]. Bioorg Med Chem,2007, 15: 1947-1958.
[5] 周大铮, 易杨华, 毛士龙,等. 香榧假种皮中的木脂素成分[J]. 药学学报, 2004, 39(4): 269-271.
[6] 周大铮. 香榧中抗艾滋病病毒活性先导化合物的研究和结构修饰[D]. 第二军医大学, 2004.
[7] Wang TT, Liu J,LU ZL,etal. Efficient and mild synthesis of highly substituted 2,5-dihydro-furan and furan derivativesviastepwise reaction[J].Tetrahedron, 2011, 67(19): 3476-3482.
猜你喜欢
浙江化工(2022年8期)2022-09-05
磷肥与复肥(2022年5期)2022-06-18
化工设计通讯(2020年7期)2020-07-25
有机氟工业(2020年2期)2020-07-04
山东化工(2020年9期)2020-06-01
农药科学与管理(2019年8期)2019-11-23
浙江化工(2015年4期)2015-11-28
火炸药学报(2015年6期)2015-03-08
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
江西理工大学学报(2013年1期)2013-03-20
