
Density functional theory study of H,C and O chemisorption on UN(001)and(111)surfaces∗
2014-08-05 09:13:24LIRuSong李如松HEBin何彬XUPeng许鹏WANGFei王飞andMAWenYan马文彦
Nuclear Science and Techniques 2014年5期
关键词:王飞
LI Ru-Song(李如松),HE Bin(何彬),XU Peng(许鹏),WANG Fei(王飞),and MA Wen-Yan(马文彦)
1Xi’an Research Institute of Hi-Tech,Xi’an 710025,China
Density functional theory study of H,C and O chemisorption on UN(001)and(111)surfaces∗
LI Ru-Song(李如松),1,†HE Bin(何彬),1XU Peng(许鹏),1WANG Fei(王飞),1and MA Wen-Yan(马文彦)1
1Xi’an Research Institute of Hi-Tech,Xi’an 710025,China
We performed density functional theory calculations of H,C,and O chemisorption on the UN(001)and (111)surfaces using the generalized gradient approximation(GGA)and the HubbardUparameter and revised Perdew-Burke-Ernzerhof(RPBE)exchange-correlation functional at non-spin polarized level with the periodic slab model.Chemisorption energies vs.distance of molecules from UN(001)and UN(111)surfaces have been optimized for four symmetrical chemisorption sites,respectively.The results show that the Hollow,N-top,and Hollow adsorption sites are the most stable sites for H,C,and O atoms with chemisorption energies of 13.06, 25.50 and 27.34kJ/mol for UN(001)surface,respectively.From the point of adsorbent(UN(001)and UN(111) surfaces in this paper),interaction of O with the chemisorbed surface is of the maximum magnitude,then C and H,which are in agreement with electronegativities of individual atoms.For the UN(001)surface,U-N bond lengths change relatively little(<9%)as a result of H chemisorption,however C and O chemisorptions result in remarkable changes for U-N bond lengths in interlayer(>10%).Electronic structure calculations indicate that Bridge position is equivalent with Hollow position,and the most stable chemisorption position for H,C, and O atoms are all Bridge(or Hollow)position for the UN(111)surface.Calculated electronic density of states(DOSs)demonstrate electronic charge transfer betweens,porbitals in chemisorbed atoms and U 6d,5forbitals.
Chemisorption,Density functional theory,Relaxation,Density of states
I.INTRODUCTION
Uranium nitrides are considered as promising fuel for the fast nuclear Generation IV reactors.Compared to many uranium and plutonium oxide nuclear fuels,uranium nitrides have several advantages[1,2],such as higher thermal conductivity,melting point,metal density,and smaller lattice constant.Many research groups have identified an important role of UN in anti-corrosion application since the 1960s[3–5],and investigations on uranium nitride compounds renew great attention due to enviormental pollution and increasing interest for development of new nuclear fuels.The bulk properties of actinide nitrides have been investigated in these works,especially the elastic and magnetic properties.
Contrary to a number of available experimental data,some theoretical work have paid much attention on the pure and defective UN,physical properties of various defects(such as vacancy,Oimpurities,grainboundary)[6–13].Basicbulkproperties have been considered in these studies for uranium nitrides with emphasis on elastic and magnetic properties[14–18].Petitet al.[9]have clarified the partial localization of 5felectrons in UN and reproduced the experimental total magnetic moment.In Ref.[7],the all-electron relativistic spin-polarized DFT calculations were performed to evaluate the total energies,optimized geometries,and electronic and thermodynamic properties of perfect stoichiometric UN and UN2single crystals.However,high reactivities of uraniumnitrides with hydrogen,carbon,and oxygen at ambient atmosphere can affect the fuel fabrication process and fuel performance[1].Experimental studies also clearly showed that oxygen in contact with the surface of uranium mononitride can result in growth of the oxide compound and,in initial stages,can lead to the formation of a surface layer structurally similar to oxynitrides UOxNy[19].
To predict UN fuel performance under different operating conditions and understand the material properties at the microscopic scale,it is crucial to investigate the surface properties and chemisorption process based on electronic structure calculations.However,a few papers have focused on the chemisorption behaviors of molecules or atoms on the surface of uranium nitrides.
Only recently[19–23],some groups have simulated the reactivity of molecules/atoms with the surface of uranium nitrides.These reports have indicated that O2molecule would spontaneously dissociate after chemisorption on the UN(001)surface,then the produced O atoms exhibit a strong chemisorption behavior.
AccordingtoTasker’sanalysis,the(001)surfacemusthave the lowest surface energy for the rock-salt compounds[24]. At the same time,the(111)surface is usually the most denseplaneforface-centeredcubic(fcc)structure.Therefore, we consider two representative planes for the UN lattice–UN(001)and UN(111)surface.In this work,we perform electronic structure calculations within the framework of density functional theory(DFT)method to deeply understand chemisorption mechanisms of H,C,and O atoms on the UN(001)and(111)surfaces,shed light on the mechanisms of hydrogenization,oxidation,and cabonization of UN in the air at the electronic and atomic level,and analyse the chemical bonding of U 6d,5fstates with H 1s,C 2s,2p,and O 2s, 2pstates.
II.METHODOLOGY
Uranium mononitride(UN)has NaCl-type structure(facecentered cubic)with the experimental lattice constanta= 0.4889nm.In this paper,we use the generalized gradient approximation(GGA)+U(HubbardUparameter to represent a correction to Coulomb repulsion interaction,separatingfmanifold into lower and upper Hubbard bands and removingfdegrees of freedom from the Fermi level,U=4.0eV)within the framework of density functional theory(DFT)and revised Perdew-Burke-Ernzerhof(RPBE) exchange-correlation functional with a periodicthree-layer slab model and single-sided chemisorption mode (an atom is placed on one side of the slab model,namely 0.5ML adsorptivity)to simulate chemisorption behaviors of H,C,and O atoms on UN(001)and(111)surfaces in all calculations.A vacuum layer of 2.0nm is added to a unit cell of the layers.The vacuum height test will be described in Sec.III in detail.We do not yet consider fully relativistic effects instead by using the scalar-relativistic approach.However,all other relativistic kinematic effects,such as massvelocity,Darwin,and higher order terms are retained.Wang and Hayet al.[25,26]have found that one can adequately describe the electronic and geometric properties of actinide complexes without treating spin-orbit effects explicitly,since we areinterestedin the chemisorptionenergies,definedasthe difference in total energies.Therefore,we expect the inclusion of other relativistic effects,such as spin-orbit coupling, spin polarization,and orbital polarization effects,will not alter the main qualitative and quantitative conclusions of our work,as discussed in other works[27].
The outer fourteen electrons(6s26p65f66d17s2)of the U atom are treated as the valence electrons and the remaining seventy-eight electrons are treated as the core electrons.DFT semi-core pseudopotentials(DSPP)and a double numerical basis set with polarization functions(DNP)have been used to treatthecoreelectronsandthevalenceelectrons,respectively. All electron basis sets are used for H,C,N,and O atoms.A 8×8×2 Monkhorst-Pack k-point mesh is applied for the Brillouin zone(BZ)integration.The convergence of a selfconsistent field(SCF)is less than 1.0×10−5eV/atom.A plane-wave cutoff energy is fixed atEcut=500eV,which is enough for convergence of chemisorption calculations.Nonmagnetic configuration is appropriate for the light actinide U element from the point of total energy,so U 5felectrons are in the delocalized 5f3electronic configuration in the present work.
Single atom,one per unit cell,is allowed to approach the UN(001)surface along four different symmetrical positions, namely(I)on the middle of the two nearest U atoms(Bridge position),(II)the adsorption atom sees a U atom located on the layer directly below the surface hollow site(Hollow position),(III)directly on top of a U atom(U-top position),and (IV)directly on top of a N atom(N-top position),as shown in Fig.1,whereas only the former three symmetrical chemisorption positions exist for the UN(111)surface(not shown here). Chemisorption energyECis optimized with respect to the heightRof the chemisorbed atom above the surface,and is given by[28]
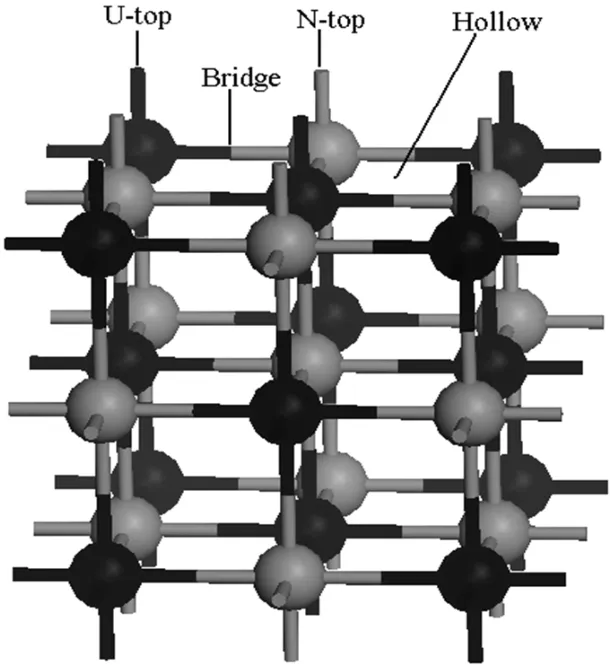
Fig.1.UN(001)surface has four symmetrical chemisorption positions,namely Bridge,Hollow,U-top and N-top,whereas only the former three symmetrical chemisorption positions exist for UN(111) surface.
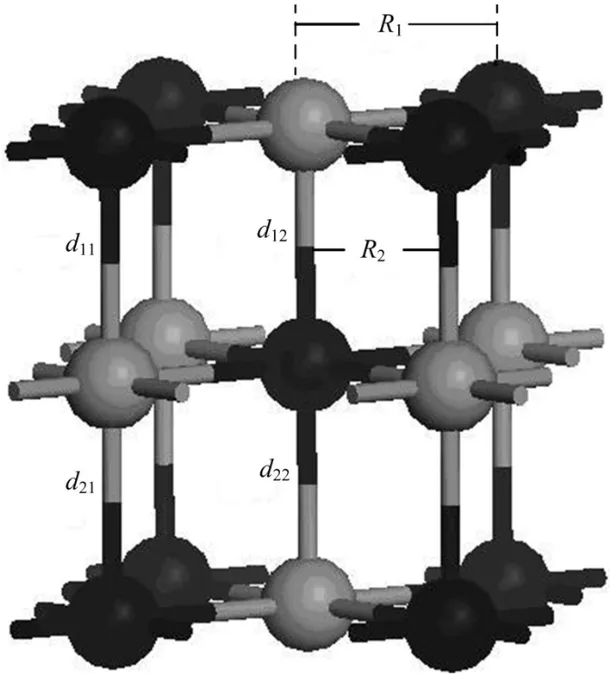
Fig.2.A configuration model for relaxation calculation.Ri(i=1, 2)anddij(i=1,2,j=1,2)represent U-N bond length in the intralayer and interlayer,respectively.
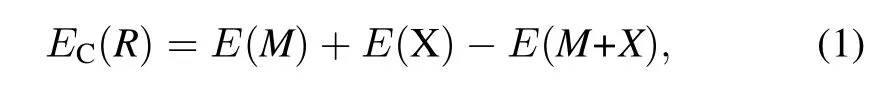
whereE(M)is total energy of a bare UN(001)or UN(111) slab,E(X)is the total energy of the isolated atom in reference crystal structure,andE(M+X)is the total energy of the entire chemisorption system.
The relative change for bond length is utilized to describe thechangeforU-Nbondlengthasaresultofatomchemisorption
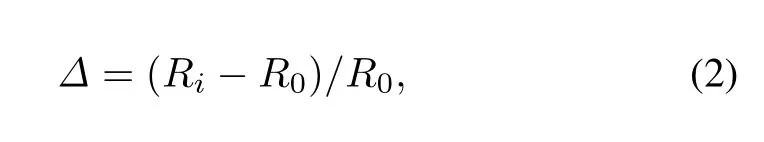
whereRiis the bond length for the central N atom on the UN(001)surface and theithU atom,R0is the original U-N bond length,andΔdenotes the relative change forRi.The serial numbersiare shown in Fig.2.
III.CONFIGURATIONS FOR UN(001)AND(111) SURFACES
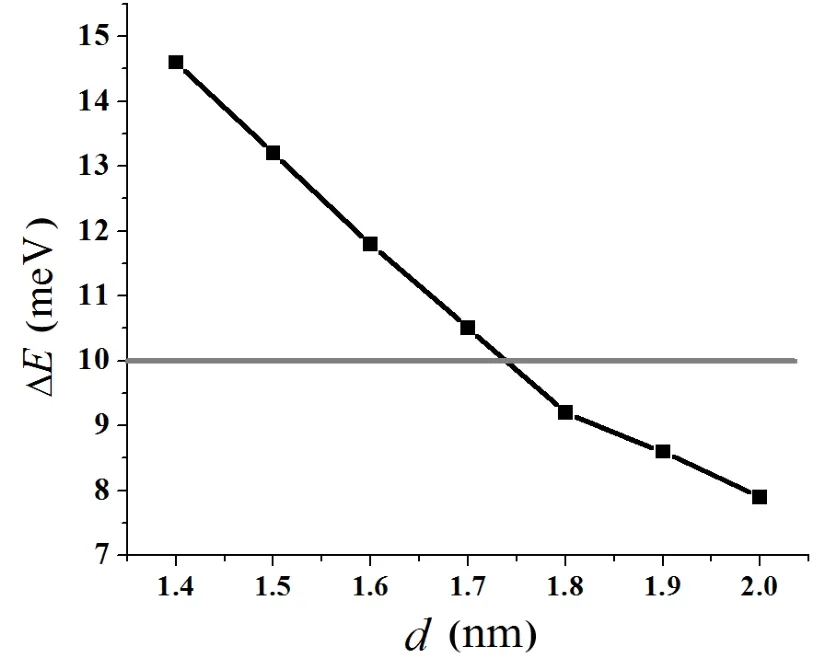
Fig.3.Convergence test of the vacuum thickness for the UN(001) surface.

Fig.4.A calculation model for the UN(001)surface.
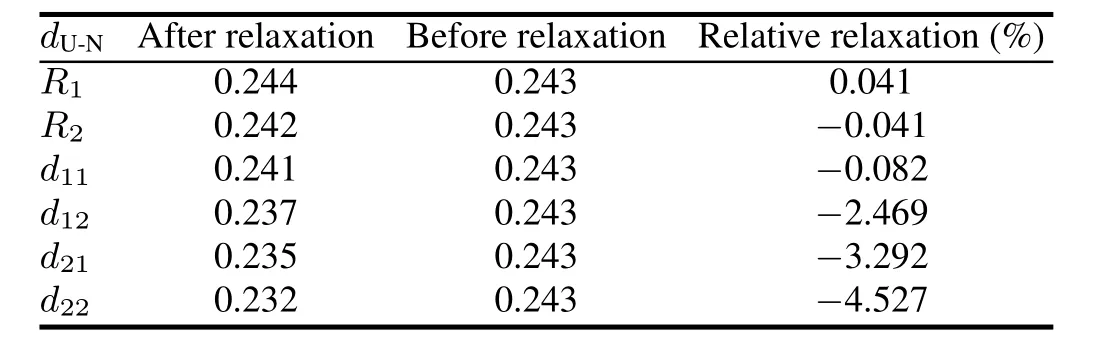
TABLE 1.Calculated results for configuration relaxation.Ri(i= 1,2,in nm)anddij(i=1,2,j=1,2,in nm)represent U-N bond length in the intralayer and interlayer,respectively
To test the validity of the computational parameters,we first cleave the UN(001)surface,then check the convergence of total energy of the UN(001)surface with different vacuum thicknesses.We consider a vacuum thickness test to be convergent as long as change for total energy ΔEis less than 10meV.Test results indicate that the total energy of a system is convergent when vacuum thickness is larger than 1.8nm (Fig.3).Therefore,we add a vacuum layer of 2.0nm onto the unit cell of the periodic slab in order to reduce the influence of the boundary condition on the computation procedure.A model for the UN(001)surface is plotted in Fig.4. face atoms to relax,reconstruct,find a new equilibrium site, and finally lower the total energy of the system.Moreover, this relaxation behavior may also change U-N bond length. A configuration model for relaxation calculation is shown in Fig.2.In our work,relaxation is defined as relative change of U-N bond length,and the calculation result is listed in Table 1.From this table,we can see that the relative relaxationisrelativelysmall(themaximumvalueatmost4.527%), so we fix the atoms in two low-lying layer in the following section,otherwise particularly declaration.For clarity, the configuration model and the relaxation calculation for the UN(111)surface are not shown here.
IV.RESULTS AND DISCUSSION
The energy minimum principle demonstrates that the higher the system symmetry,the lower the system energy, and the more stable the system.Therefore,we prefer an atom to be chemisorbed onto the high symmetrical position in the crystal surface.The UN crystal has fcc structure and several high symmetrical chemisorption positions exist on the crystal surface.As discussed above,we consider four representative positions in this work,namely Bridge,Hollow,U-top, and N-top.The chemisorbed atom is directly placed on the top of individual positions to study chemisorption behavior of the atom on these positions,as shown in Fig.1.Chemisorption parameters not only include position,but also the orientation and height of the atom from the UN(001)and(111) surfaces,where chemisorption orientation is associated with the molecule structure and chemisorption height mainly depends on stability of the system.
A.H atom
We de fi ne chemisorption heighthas the nearest distance for an H atom from UN(001)and UN(111)surfaces,and express it in terms of fractional coordinates.We optimize the H-UN(001)/UN(111)system with the minimum total energies(Table 2).The results for H chemisorption energies and Mulliken charges of individual configurations are listed in Table 3.As shown in Table 2,chemisorption heights of Bridge position are the same as Hollow position,and chemisorption energy differences are within several MeV.We think Bridge position to be equivalent with Hollow position.Toverify this statement,we still consider Bridge,Hollow,and U-Top positions for C and O chemisorptions.The most stable position for H chemisorptions on the UN(001)and UN(111)surfaces are Hollow position(chemisorption energy 13.06kJ/mol)and Bridge/Hollow position(chemisorption energy 11.532kJ/mol),respectively.
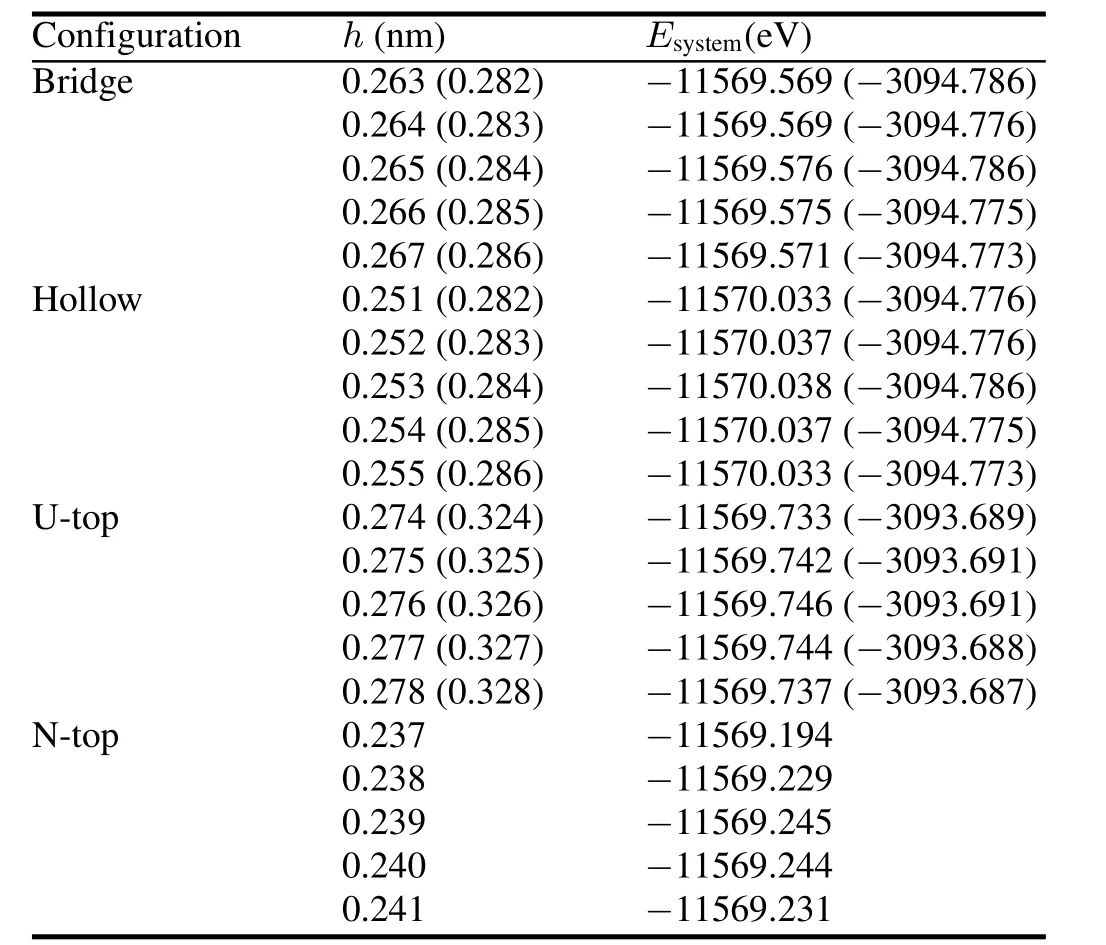
TABLE 2.Total energies of H-UN(001)/UN(111)systems with different chemisorption positions and heights.Numerical values for H-UN(111)system are listed in parentheses.Meanings for Bridge, Hollow,U-top and N-Top configurations are discussed in the text

TABLE 3.Chemisorption configurations,chemisorption energiesEC(inkJ/mol)andMullikenchargesQ(ine)forHatom.Numerical values for H-UN(111)system are listed in parentheses.Meanings for Bridge,Hollow,U-top and N-Top configurations are discussed in the text
To further understand interaction of H atoms with the UN(001)surface,we analyze the projected density of states (PDOS)before and after H chemisorption in terms of electronic structure calculations,as shown in Fig.5(a)–5(c).In fact,the ground state valence electronic configurations of U and N atoms are 5f36d17s2and 2s22p3,respectively.From the point of ionic bonding behavior,the U6dand 7selectrons fill the N2pstates and the three U5felectrons form the highest occupied molecular orbital(HOMO)[29].The characteristic peak of H 1sPDOS in the energy range of 17.157eV and 20.271eV shifts towards lower energy band induced by H chemisorption(Fig.5(a))and expands its peak area,indicating that H atoms gain electrons as a result of chemisorption,which is in agreement with the Mulliken charge analysis (the third row in Table 3).We neglect others,porbitals inU atoms because 6d,5forbitals dominate the structural and electronic properties of the U element.
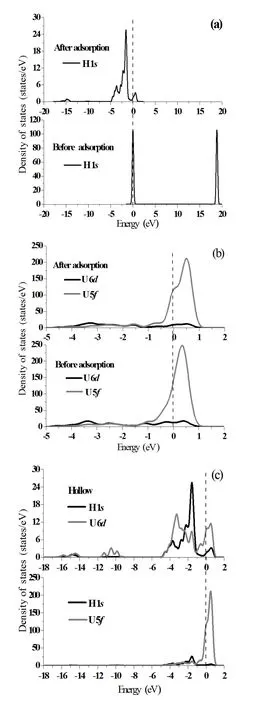
Fig.5.Projected density of states(PDOS)of H 1sorbital(a),U 6d,5forbitals(b)and H 1s,U 6d,5forbitals(c)before and after H chemisorption on the Hollow position of UN(001)surface.The Fermi energy(dash line)stands at 0eV.
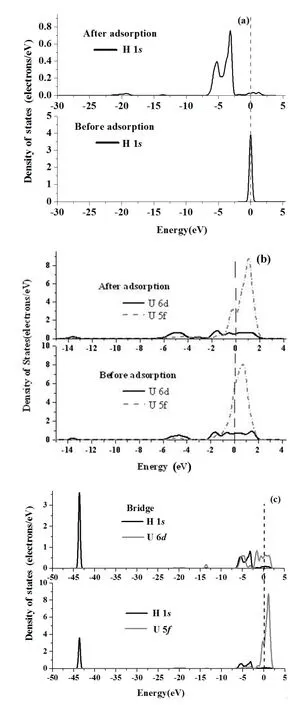
Fig.6.Projected density of states(PDOS)of H 1sorbital(a),U 6d,5forbitals(b)and H 1s,U 6d,5forbitals(c)before and after H chemisorption on the Bridge or Hollow positions of UN(111) surface.The Fermi energy(dash line)stands at 0eV.
We can see that H chemisorption causes the shape of U 5fPDOS with energy of−1.026eV~0eV to slightly change and peak position of U 5fPDOS shifts from 0.353eV to 0.502eV(Fig.5(b)).From this plot we can see that peakvalue decreases from 247.337states/eV to 211.083states/eV and peak area of U 6dPDOS obviously diminishes with energies of−1.042eV and 1.515eV,which show that U 6d,5forbitals both lose electrons,also in agreement with the Mulliken charge analysis(Table 3).Fig.5(c))depicts H 1sand U 6d,5fPDOS.H 1sorbitals remarkably overlap with U 6dorbitals in the energy of−5.043eV and 1.161eV.However, H 1sorbitals almost separate from U 6forbitals with energy intervals of−4.152eV and−1.241eV.It is known that the larger the overlapping area of PDOS,the higher the hybridized bonding.Therefore,the H atom will hybridize with the U atom as a consequence of H chemisorption.Electronic charges of U 6d,5forbitals(mainly U 5forbitals)transfer to the H 1sorbital,which is also consistent with the Mulliken charge analysis(Table 3).
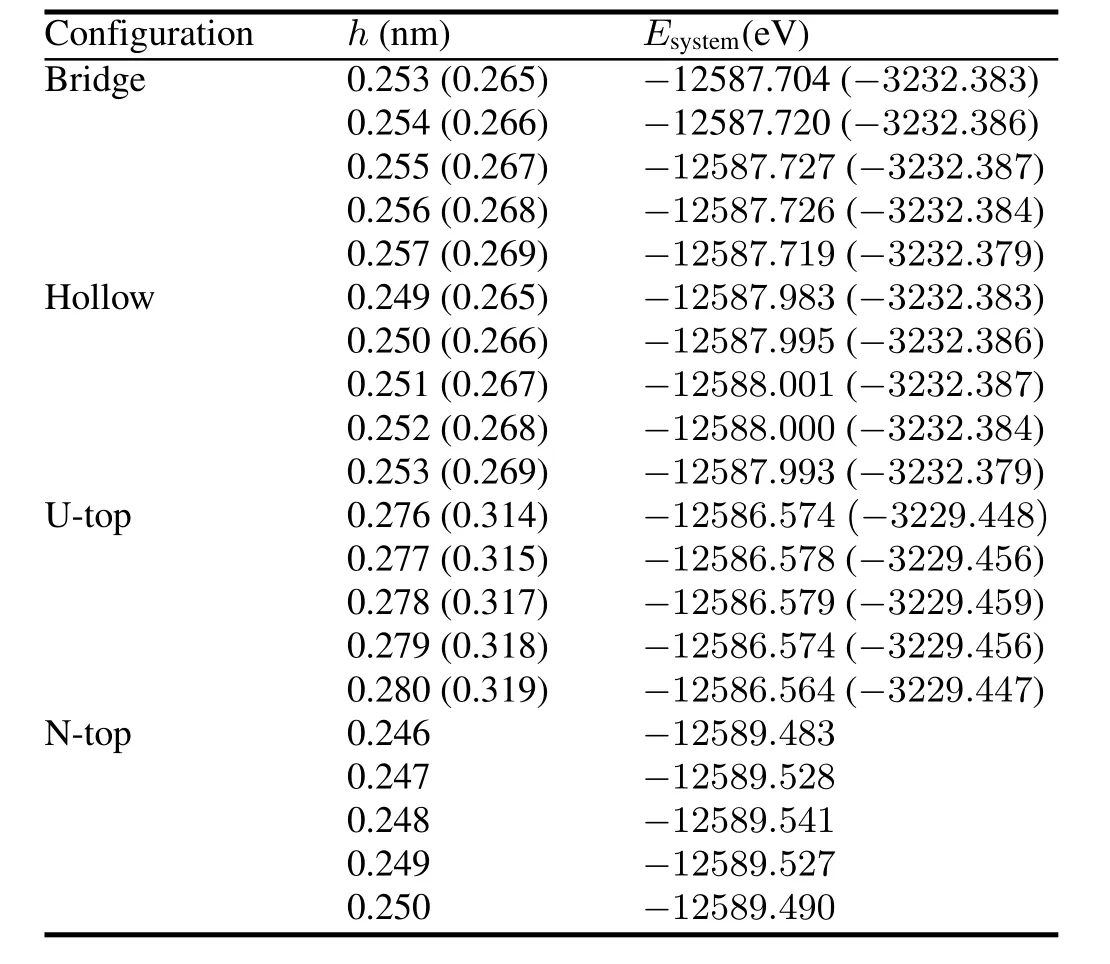
TABLE 4.Total energies of C-UN(001)/UN(111)systems with different chemisorption positions and heights.Numerical values for C-UN(111)system are listed in parentheses.Meanings for Bridge, Hollow,U-top and N-Top configurations are discussed in the text
Similarly,PDOSs before and after H chemisorption in Bridge or Hollow positions for the UN(111)surface are shown in Figs.6(a)–6(c).H chemisorption induces an H 1speak around the Fermi level to the energy band in the energy range of−7.374eV to−2.502eV(Fig.6(a)).However, H chemisorption has little effect on U 6d,5fPDOSs.U 6dstates clearly hybridize with U 5fstates between−2.215eV and 2.00eV,as shown in Fig.6(b).H 1sstates clearly overlap with U 6d,5fstates(Fig.6(c)),and acquire electrons from U atom,which is consistent with Mulliken analysis(Table 3).
B.C atom
Previous report[2]has shown that U carbides can effectively retard corrosion of U metal.The crystal structure of UN is the same as that of UC(NaCl type,face-centered cubic structure)and the lattice constant is also very close to the latter(lattice constants of UN and UC are 0.4965nm and 0.4889nm,respectively).Therefore,we wonder whether chemisorption behavior of a C atom on UN(001) and UN(111)surfaces is also similar with that of a U monocarbide.

TABLE 5.Chemisorption configurations,chemisorption energiesEC(in kJ/mol)and Mulliken chargesQ(ine)for C atom.Numerical valuesforC-UN(111)systemarelistedinparentheses.Meaningsfor Bridge,Hollow,U-topandN-Topconfigurationsarediscussedinthe text
Next we perform electronic structure calculations of total energy for the C-UN(001)/UN(111)systems with different chemisorption positions and heights using the method discussed above and relax different configurations to find the optimal chemisorption positions and heights.The total energy results of the C-UN(001)/UN(111)systems are listed in Table 4.According to the chemisorption energy formula (Eq.(1)),a configuration with the maximum chemisorption energy is the most stable configuration for chemisorption on the UN(001)and UN(111)surface.Therefore,we optimize configurations with the minimum total energy for different chemisorption positions and the optimal configuration is the most stable configuration for certain chemisorption positions.
ThechemisorptionenergyandMullikenchargeanalysisresults are presented in Table 5.From this table we can see that chemisorption energies reach the maximum value when the C atom chemisorbs or at the Bridge and N-top positions of the UN(001)surface(chemisorption energies are 25.513kJ/mol and 25.502kJ/mol,respectively).The chemisorption energy of the Bridge configuration is very close to that of the N-top configuration.The relaxtion calcultion shows that the former is unstable,and will transform to the latter.Therefore,the most stable configuration for C chemisorption on the UN(001)surface is N-top configuration.Similarly,relaxation result shows that the most stable configuration for C chemisorption on the UN(111)surface is the Bridge or Hollow configurations.
To further understand the interactions of C with the UN(001)and(111)surfaces,we analyze PDOS before and after C chemisorption in terms of electronic structure calculations,as shown in Figs.7(a)–7(d)and Figs.8(a)–8(d). C 2sand 2pPDOSs shift towards a lower energy band after chemisorption(Fig.7(a)),and the peak area of C 2sand 2pPDOSs obviously increase,indicating that C atoms gain electrons from the adsorbate(UN(001)surface in the present work)as a result of chemisorption,which is in agreement with the Mulliken charge analysis(Table 5).U 6d,5fPDOSs before and after chemisorption are plotted in Fig.7(b).C chemisorption has a negligible effect on U 6dPDOS,however,the peak value of the U 6dPDOS decreases(Fig.7(b)), which shows that U 6dorbitals lose electrons.At the same time,C chemisorption also has a negligible effect on the U 5fPDOS in the energy interval of 0eV~1.520eV.However,a new characteristic peak appears in the energy range of−1.0eV~0.50eV,showing that U 5forbitals hybridize with C 2sor 2porbitals.As shown in Fig.7(b),peak position of U 5fPDOSshiftsfrom0.508eVto0.572eVandpeakvaluedecreases from 241.746states/eV to 227.908states/eV,indicating that U 5forbitals also lose electrons.Figs.7(c)–7(d))depict interactions of U 6d,5forbitals with C 2s,2porbitals.C 2sorbitals and U 6dorbitals produce a remarkable hybridization/mixing peak(Fig.7(c)),while the C 2sPDOS almost separates from the U 5fPDOS.However,C 2porbitals obviously overlap with U 6d,5forbitals(Fig.7(d)),showing that C 2porbitals hybrid with U 6d,5forbitals to form a covalent bond,which is in sharp contrast with Fig.7(c).Therefore, electronic charges of U 6dorbitals transfer to C 2s,2porbitals as a result of C chemisorption on the UN(001)surface and U 5forbitals transfer to C 2porbitals,these results are consistent with the Mulliken charge analysis(Table 5).
As shown in Fig.8(a),C chemisorption widens the C 2ppeak around the Fermi level,and new C 2sstates appear in the energy interval of−5.241eV~−2.632eV to−21.352eV~−18.471eV(Fig.8(a)).However,C chemisorption also has a negligible effecton U 6d,5fPDOSs (Fig.8(b)).C 2sstates slightly overlap with U 6d,5fstates (Fig.8(c)).However,C 2pstates obviously hybridize with U 6d,5fstates(Fig.8(d))and acquire a majority of electrons (about 2/3)from the U atom,which is consistent with Mulliken analysis(Table 5).
C.O atom
Previous report[30]has shown that H2O chemisorption on U metal will result in the formation of UO2passivation film on the metal surface and that this film can prevent U metal from further oxidization.Therefore,investigation of O chemisorption on the UN surface may be helpful for understanding anti-corrosion mechanisms of U metal because UN passivation fi lm is also a well-known corrosion-resistant material.
According to the minimum energy principle,the total energy of a system would be the minimum value for H2O chemisorption on the optimal position and the system would be the most stable one.Therefore,we first perform total energy calculations for O-UN(001)/UN(111)systems with different chemisorption positions and heights,and then optimize several systems with the minimum energies(Table 6).The calculation results are listed in Table 7.From this table,we can see that the most stable configuration for O chemisorption on the UN(001)surface is Hollow configuration.O chemisorption induces the UN(001)surface to reconstruct,where U atoms move outwards and N atoms move towards the matrix,thereby causing U-N bond length to change.The maximum relative change for U-N bond length reaches 15.79%,indicating that O atom strongly interacts with the UN(001)surface(not shown here).

Fig.7.Projected density of states(PDOS)of C 2s,2porbital(a),U 6d,5forbital(b),C 2s,U 6d,5forbital(c)and C 2p,U 6d,5forbital (d)before and after C chemisorption on N-top position of the UN(001)surface.The Fermi energy(dash line)stands at 0eV.
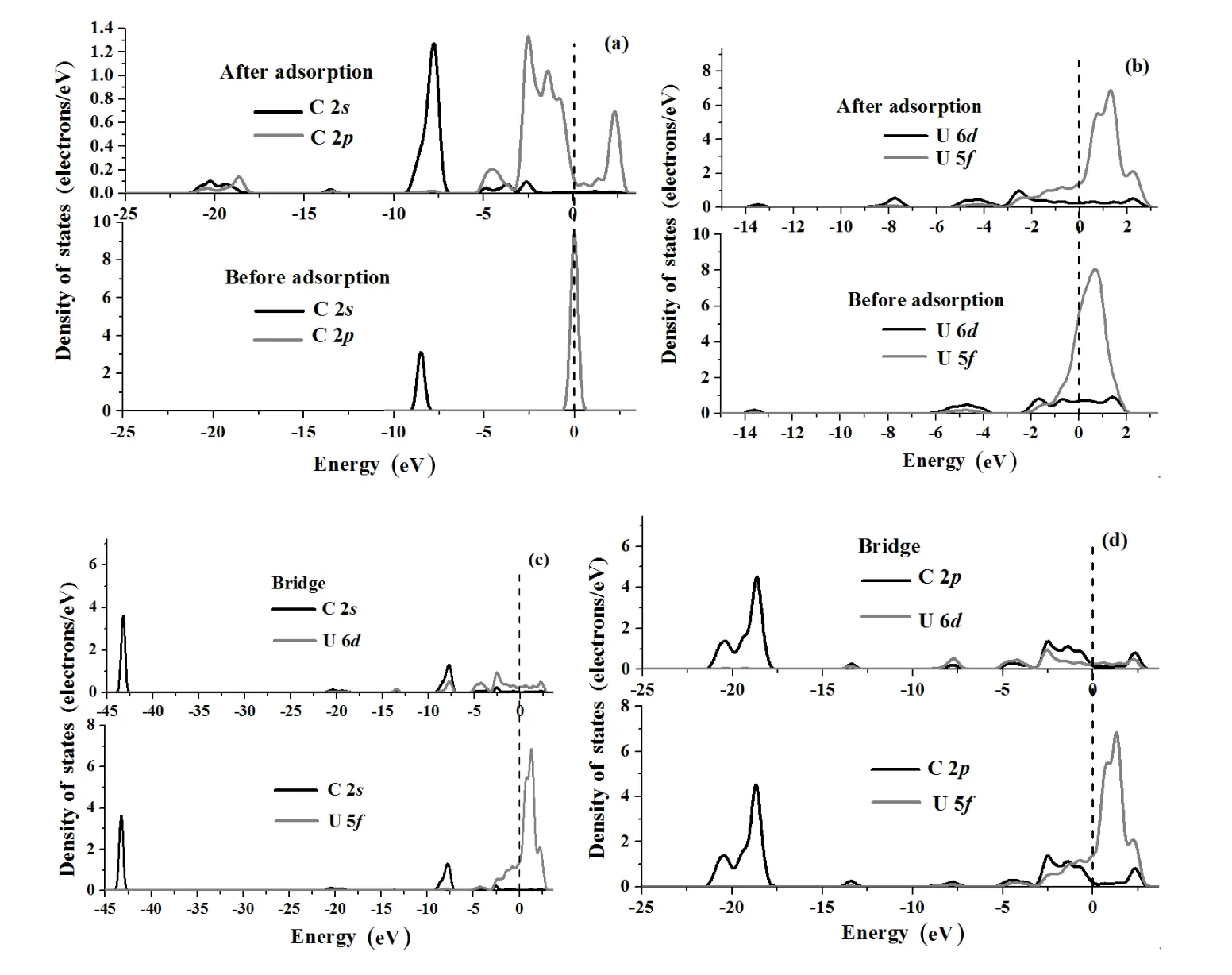
Fig.8.Projected density of states(PDOS)of C 2s,2porbital(a),U 6d,5forbital(b),C 2s,U 6d,5forbital(c)and C 2p,U 6d,5forbital (d)before and after C chemisorption on Bridge or Hollow positions of the UN(111)surface.The Fermi energy(dash line)stands at 0eV.

Fig.9.Projected density of states(PDOS)of H 1s,O 2s,2porbital(a),U 6d,5forbital(b),O 2s,U 6d,5forbital(c)and O 2p,U 6d,5forbital(d)before and after O2chemisorption on the BU position of the UN(001)surface.The Fermi energy(dash line)stands at 0eV.
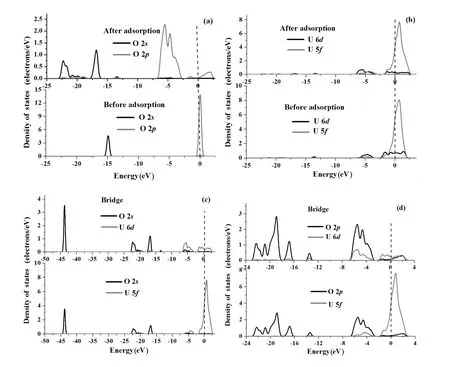
Fig.10.Projected density of states(PDOS)of H 1s,O 2s,2porbital(a),U 6d,5forbital(b),O 2s,U 6d,5forbital(c)and O 2p,U 6d,5forbital(d)before and after H2O chemisorption on Bridge or Hollow positions of the UN(111)surface.The Fermi energy(dash line)stands at 0eV.
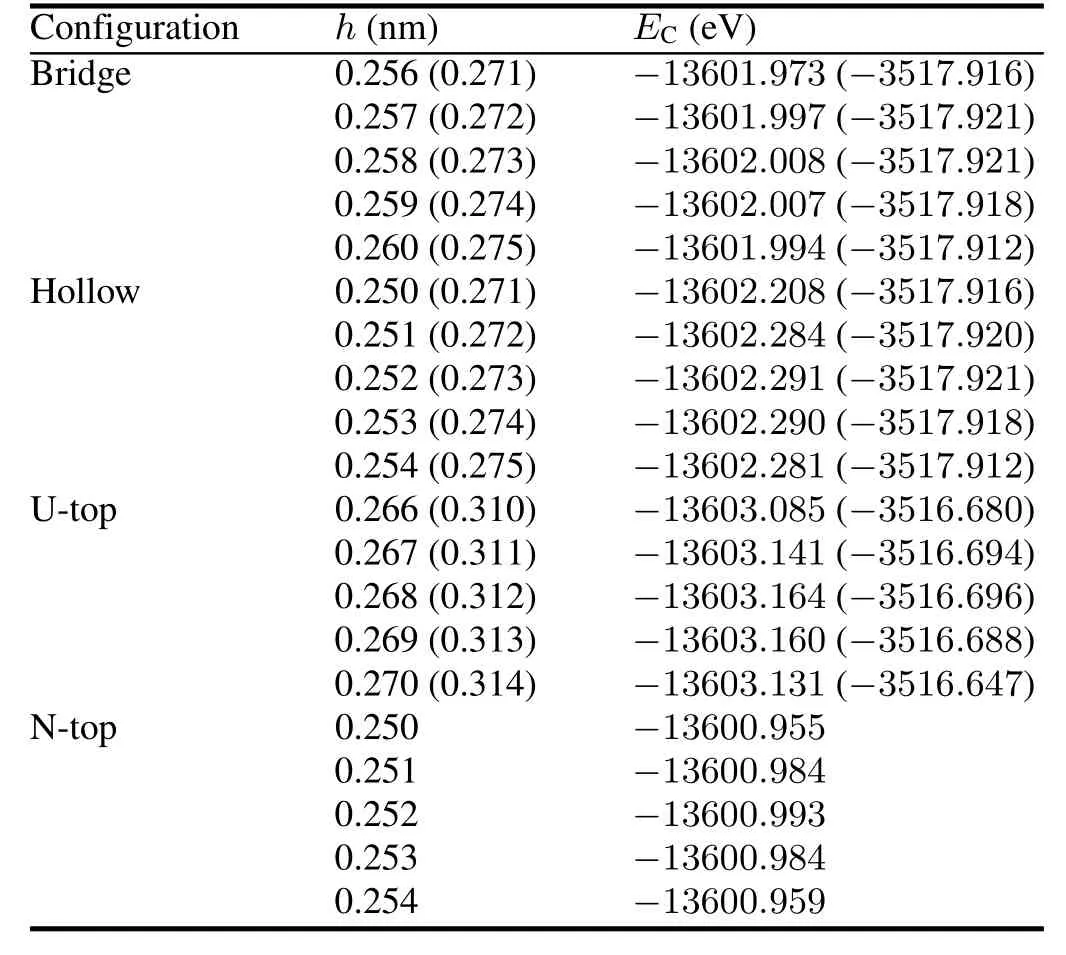
TABLE 6.Chemisorption heights and chemisorption energiesEC(in kJ/mol)for O atom on UN(001)and UN(111)surafces.Numerical values for O-UN(111)system are listed in parentheses.Meanings for Bridge,Hollow,U-top and N-Top configurations are discussed in the text

TABLE 7.Chemisorption energiesEC(in kJ/mol)and Mulliken chargesQ(ine)for O atom
V.CONCLUSION
We also performed electronic structure calculations for O chemisorptionontheUN(001)andUN(111)surfaces,anddepictedthePDOSbeforeandafterchemisorptioninFigs.9(a)–9(d)and Figs.10(a)–10(d).C 2sand 2pPDOSs shift towards a lower energy band after chemisorption(Fig.9(a)) and the peak area of the C 2pPDOS increases,indicating that C atoms gain electrons from the UN(001)as a result of chemisorption,which is in agreement with the Mulliken charge analysis(the third row in Table 7).Peak areas of the U 6d,5fPDOS in the energy of−1.5eV~1.3eV diminish,which shows that U 6d,5felectrons transfer to the O 2sor 2porbitals.However,peak areas of the U 6d,5fPDOS in the energy of−5.0eV~−1.50eV increase,indicating that U 6d,5forbitals may hybridize with O 2sor 2porbitals(Fig.9(b)).The peak position of U 5fPDOS shifts from0.354eVto0.418eV,andthepeakvaluedecreasesfrom 247.361states/eV to 237.268states/eV,implying that U 5forbitals lose electrons.U 6dorbitals and O 2sorbitals form a relatively small overlapping peak,while the U 5fPDOS is almost separate from the O 2sPDOS(Fig.9(c)).However,O 2pPDOSobviouslyoverlapwithU6d,5fPDOSs(Fig.9(d)), particularly the U 6dPDOS,which is in sharp contrast with Fig.9(c).It is shown that the smaller the overlapping area,the weaker the chemical bonding and the lower the transferred charge.Therefore,electronic charges of U 6dorbitals transfer to O 2sand 2porbitals(mainly O 2porbitals)and U 5forbitals transfer to O 2porbitals,which is in agreement with the Mulliken charge analysis(Table 7).
From Fig.10(a),we can see that O chemisorption shifts the O 2s,2ppeak to a lower energy,and widens O 2pPDOS. However,O chemisorption has an ignorable effect on U 6d, 5fPDOSs(Fig.10(b)).O 2sstates slightly overlap with U 6d,5fstates(Fig.10(c)).However,O 2pstates remarkably hybridize with U 6d,5fstates(Fig.10(d))and acquire electrons from the U atom,which is consistent with Mulliken analysis(Table 7).
In the present work,we performed density functional theory calculations for H,C,and O chemisorptions on UN(001) and UN(111)surfaces using the generalized gradient approximation(GGA)and revised Perdew-Burke-Ernzerhof(RPBE) exchange-correlation functional at a non-spin polarized level with a periodic slab model.The results show that Hollow, N-top,and Hollow chemisorption sites are the most stable sites for H,C,and O atoms chemisorbed on the UN(001) suraface,respectively.The calculated electronic density of states(DOS)demonstrate electronic charge transfer betweens,porbitals in chemisorbed atoms and U 6d,5forbitals and transferred electronic charges agree with the Mulliken charge analysis.Electronic structure calculations of H,C and O atom chemisorption on Bridge,Hollow,and U-Top positions of the UN(111)surface indicate that Bridge position is equivalent with Hollow position,two symmetrical chemisorption positions exist on UN(111)surface,namely Bridge(or Hollow) and U-Top,and the most stable chemisorption position for H, C,and O atoms is Bridge(or Hollow)position.
In the future,we plan to investigate the chemisorption of other atoms or molecules on the surface of actinide compounds,especially promising nuclear fuels such as Pu and U compounds,providing corresponding anti-corrosion techniques,and improving operational life and ef fi ciency for nuclear fuel.
ACKNOWLEDGMENTS
We would like to thank our colleagues for their helpful comments and suggestions on this manuscript.
[1]Kotomin E A and Mastrikov Yu A.J Nucl Mater,2008,377: 492–495.
[2]Bocharov D,Gryaznov D,Zhukovskii Yu F,et al.Surf Sci, 2011,605:396–400.
[3]Shuai M B,Hu H R,Wang X,et al.J Mol Struc-Theochem, 2011,536:269–276.
[4]Dholabhai P P and Ray A K.J Alloy Compd,2007,444-445: 356–362.
[5]Burns C J.Science,2005,309:1823–1824.
[6]Shibata H,Tsuru T,Hirata M,et al.J Nucl Mater,2010,401: 113–117.
[7]Weck P F,Kim E,BalakrishnanN,etal.Chem Phys Lett,2007,443:82–86.
[8]Evarestov R A,Bandura A V,Losev M V,et al.J Comput Chem,2008,29:2079–2087.
[9]Petit L,Svane A,Szotek Z,et al.Phys Rev B,2009,80: 045124.
[10]Sunder S and Miller N H.J Alloy Compd,1998,271:568–572.
[11]Brooks M S and Glotzel D.Physica B,1980,102:51–58.
[12]Brooks M S.J Phys F Met Phys,1984,14:639–640.
[13]Sedmidubsky D,Konings R J,Novak P.J Nucl Mater,2005,344:40–44.
[14]Kotomin E A,Grimes R W,Mastrikov Y,et al.J Phys-Condens Mat,2007,19:106208.
[15]Evarestov R A,Losev M V,Panin A I,et al.Phys Status Solidi B,2008,245:114–122.
[16]Czekata M S,Talik E,Troc R,et al.Phys Rev B,2007,76: 144426.
[17]Fynn R A and Ray A K.Phys Rev B,2007,76:115101.
[18]Savrasov S Y and Kotliar G.Phys Rev Lett,2000,84:3670–3673.
[19]Reihl B,Hollinger G,Himpsel F J.Phys Rev B,1983,28: 1490–1494.
[20]Ritchie A G.J Nucl Mater,1981,102:170–182.
[21]Winer K,Colmenares C A,Smith R L.Surf Sci,1987,183: 67–99.
[22]Zhukovskii Yu F,Bocharov D,Kotomin E A,et al.Surf Sci, 2009,603:50–53.
[23]Zhukovskii Yu F,Bocharov D,Kotomin E A.J Nucl Mater, 2009,393:504–507.
[24]Tasker P W.J Phys C Solid State,1979,12:4977–4984.
[25]Wang Y and Sun Y.J Phys-Condens Mat,2000,12:L311–L316.
[26]Hay P J and Martin R L.J Chem Phys,1998,109:3875–3881.
[27]Delley B.Phys Rev B,2002,66:155125.
[28]Huda M N and Ray A K.Physica B,2004,352:5–17.
[29]Marutzk M,Barkow U,Schoenes J,et al.J Magn Magn Mater, 2006,299:225–230.
[30]Hodkin E N.J Nucl Mater,1977,67:171–180.
10.13538/j.1001-8042/nst.25.050502
(Received March 28,2014;accepted in revised form June 27,2014;published online October 4,2014)
∗SupportedbyNationalNaturalScienceFoundationofChina (Nos.51401237,51271198 and 11474358)and Self-Topics Fund of Xi’an Research Institute of High Technology(Nos.2014QNJJ018 and YX2012cxpy06)
†Corresponding author,rusong231@126.com
猜你喜欢
证券市场周刊(2024年23期)2024-07-13 13:58:07
证券市场周刊(2024年4期)2024-02-02 19:20:53
证券市场周刊(2024年4期)2024-02-02 19:20:53
证券市场周刊(2024年3期)2024-01-25 06:36:47
证券市场周刊(2024年3期)2024-01-25 06:36:47
证券市场周刊(2024年2期)2024-01-14 20:38:15
当代作家(2023年3期)2023-04-23 01:58:26
当代作家(2023年3期)2023-04-23 01:58:26
证券市场红周刊(2019年37期)2019-10-09 22:23:51
证券市场红周刊(2019年37期)2019-10-09 00:48:03
 Nuclear Science and Techniques2014年5期
Nuclear Science and Techniques2014年5期
- Nuclear Science and Techniques的其它文章
- The effect of Nb additive on Te-induced stress corrosion cracking in Ni alloy:a fi rst-principles calculation∗
- Development and testing of the code for automatic generating of multi-temperature continuous-energy neutron cross section libraries∗
- Steady thermal hydraulic characteristics of nuclear steam generatorsbased on the drift flux code model∗
- A method of estimating and subtracting the hydrogen background in the natural carbon target used in the12C+12C experiment∗
- The extraction and smoothing algorithms for γ-ray spectrum of a CdZnTe detector system
- Applying image processing method to treat digital signals∗