
高表达瞬时电位受体通道1在大鼠弥漫性轴索损伤中的作用机制
2014-07-18 11:53宋锦宁赵永林李丹东付宙锋张斌飞
西安交通大学学报(医学版) 2014年6期
张 明,宋锦宁,李 宇,赵永林,李丹东,吴 媛,付宙锋,张斌飞
(西安交通大学医学院:1.第二附属医院神经外科,陕西西安 710004;2.第一附属医院神经外科,陕西西安 710061;3.第二附属医院重症医学科,陕西西安 710004)
◇专题研究◇
高表达瞬时电位受体通道1在大鼠弥漫性轴索损伤中的作用机制
张 明1,2,宋锦宁2,李 宇2,赵永林2,李丹东2,吴 媛3,付宙锋2,张斌飞2
(西安交通大学医学院:1.第二附属医院神经外科,陕西西安 710004;2.第一附属医院神经外科,陕西西安 710061;3.第二附属医院重症医学科,陕西西安 710004)
目的 研究瞬时电位受体通道1(TRPC1)在弥漫性轴索损伤(DAI)中的作用,探讨TRPC1在DAI后神经元钙超载及髓鞘变性中的机制。方法 瞬间旋转损伤法建立大鼠DAI模型,用SKF96365干扰TRPC1通道活性后建立DAI干预组,Fura-2AM检测胞内Ca2+浓度,Western blot和免疫组化检测皮层TRPC1蛋白的动态表达,并与正常组、DMSO对照组相比较,电镜检测神经元及髓鞘改变,TUNEL检测皮层神经元凋亡。单因素方差分析数据。结果 DAI后皮层中TRPC1蛋白表达升高,在1 d达到高峰,随后逐渐下降,同时皮层神经元胞内Ca2+浓度在1 d达到峰值,髓鞘结构破坏。给予TRPC1通道阻断剂SKF96365后,皮层TRPC1蛋白表达被抑制,皮层神经元钙内流减低,髓鞘结构部分保留,神经功能评分改善。结论 DAI后TRPC1导致的钙超载是造成髓鞘变性及神经元死亡的重要原因,抑制TRPC1产生的钙内流可保护髓鞘结构完整,减轻神经元凋亡。
瞬时电位受体通道1(TRPC1);弥漫性轴索损伤;髓鞘变性;钙超载;神经元凋亡
创伤性脑损伤(traumatic brain injury, TBI)是目前国内外造成各类年龄段人群高致残率和高死亡率的首要原因,对患者及其家庭造成沉重的社会及经济压力。TBI后神经病理改变多种多样,其中最严重的是弥漫性轴索损伤(diffuse axonal injury, DAI)[1]。DAI可反映在机械负荷下头部的加速性损伤造成选择性大脑白质轴突水肿、轴索断裂等改变。由于中枢神经系统的神经元再生能力有限,且在临床中观察到轴突损伤后致残率较高,因此轴突变性的发病机制研究变得尤为重要。有研究报道轴突外Ca2+可能来源于“轴浆池”或胞外钙库,这些Ca2+离子通道被位于神经纤维节间的多分子复合物或质膜上的离子通道所调控[2-3],其过度激活可诱发钙库或钙池释放出过量Ca2+而导致神经损伤。瞬时受体电位离子通道C1(canonical transient receptor potential channel 1, TRPC1)在质膜Ca2+转运中起重要作用,而其在DAI后轴突损伤中的作用尚不明确。本研究采用瞬间旋转损伤模型建立大鼠DAI模型,初步探讨TRPC1离子通道在轴索损伤及神经元钙超载中的作用,揭示TRPC1参与DAI后神经损伤机制,寻找临床干预的新靶点。
1 材料与方法
1.1 实验动物及主要试剂、仪器 SPF级具有相同遗传背景的健康成年雄性SD大鼠70只,体质量250~300 g,由西安交通大学医学院实验动物中心提供(许可证号:SCXK(陕)08-018)。SKF96365(S7809, Sigma-Aldrich,美国);二甲基亚砜溶液(DMSO,D8418,Sigma-Aldrich,美国);细胞膜蛋白提取试剂盒(Thermo Scientific,美国);TRPC1兔单克隆抗体(Abcam,美国);NeuN(Chemicon, MAB377B,美国);β-actin鼠单克隆抗体(Santa Cruz,美国);辣根过氧化物酶标记的IgG二抗(Santa Cruz,美国);PVDF膜(Millipore,美国);TUNEL凋亡试剂盒(Promega, 美国);凝胶成像系统(JS-380A,中国);图像采集与分析系统(Leica-Q550CW,德国);透射式电子显微镜(H-600型;HITACHI,日本);SPSS 18.0统计软件(美国);脑立体定向仪(西安交通大学医学院人体解剖学教研室提供)。
1.2 实验动物分组 随机分为DAI模型组30只,SKF96365干预模型组30只,DMSO干预对照模型组5只,正常对照组5只。DAI模型组及SKF96365干预模型组各分为5个亚组,分别为:1、3、5、7、10 d组各6只。
1.3 大鼠DAI模型的建立及分组处理 采用瞬间旋转损伤法建立大鼠DAI模型[4]。大鼠术前禁食禁饮6 h,100 g/L水合氯醛腹腔注射麻醉,麻醉剂量为0.2 mL/100 g。麻醉成功后将其俯卧位固定于旋转平台,即大鼠头部由固定双侧外耳道的耳棒、头夹及前磨牙固定孔共同固定于外侧头的旋转装置,而大鼠身体则与固定平台呈30°夹角。按下旋转开关后,大鼠头部迅速旋转90°停止,此过程包含加速及减速运动。之后复位装置,重复致伤过程8次。SKF96365干预模型组使用TRPC1通道阻断剂SKF96365 5 μL溶于DMSO,终浓度为10 μmol/L,于造模前30 min经立体定向侧脑室注射(前囟后1 mm,旁开1.5 mm,深3.5 mm),其他操作与DAI组相同;DMSO干预模型组使用等量DMSO行侧脑室注射,其他操作与DAI组相同;正常对照组不予任何处理。
1.4 大鼠神经功能缺损评分 根据改良大鼠神经功能缺损评分(modified neurological severity score, mNSS)法[5]对各组大鼠进行神经行为学评分。主要包括运动实验、感觉实验、平衡木实验、反射丧失及不正常运动,其中运动实验包括提尾实验及平地运动实验,感觉实验包括放置实验和本体感觉实验,反射及不正常运动包括耳廓反射、角膜反射、惊恐反射及癫痫、肌阵挛、肌张力障碍。
1.5 HE染色、免疫组化染色及电镜标本采集 所有大鼠饲养到对应时间后,用100 g/L水合氯醛以0.5 mL/kg麻醉,剪开胸腔,剥开心包膜,暴露心脏,将下腔静脉和腹主动脉用动脉夹夹闭,剪开左心室将灌注头插入主动脉然后用动脉夹固定,剪开右心耳放出血液,用生理盐水快速将血管内的血冲洗干净,再注入40 g/L的多聚甲醛固定液500 mL灌流固定。之后取脑用石蜡包埋,并制成5 μm切片,采用HE法及SABC法进行组织化学染色并在光镜下进行观察。在处理电镜组织标本的时候,生理盐水灌注如前,再用25 mL/L的戊二醛固定液1 000 mL灌注固定,将皮层固定于电镜固定液中,待制作电镜标本。透射电镜标本:将皮层标本用电镜固定液固定,采用常规电镜标本制作方法,经清洗、脱水、浸透、包埋和修块后作超薄切片,在透射电镜下观察。
1.6 [Ca2+]i荧光检测 取大脑顶叶皮层3 mm×3 mm 剪碎后置于1.5 mL冰Hanks液中,轻轻吹打10 min,过200目滤网。低温下以1 000 r/min离心5 min,取沉淀用冰Hanks液洗2次,用含100 mL/L小牛血清的DMEM培养液制成约106个/mL的单细胞悬液备用。锥虫蓝(台盼蓝)染色镜下观察200个细胞,计数存活细胞数>95%。取细胞悬液1 mL离心弃上清后加入含100 mL/L小牛血清的Hanks液1 mL,37 ℃预温5 min后加入7.5 μL 的Fura-2AM (终浓度为6 μmol/ L);恒温振荡孵育45 min后用含100 mL/L小牛血清的Hanks液洗涤2次;另取同一细胞悬液1 mL不负载Fura-2AM作为空白对照。
荧光测定:上述单细胞悬液37 ℃复温2~3 min后在流式细胞仪上检测荧光强度。测定时激发波长为380 nm,发射波长为510 nm,分别测定F,Fmax, Fmin。F为380 nm激发波长测得的荧光强度值;Fmax为最大荧光强度值,即加入Triton X-100(终浓度为1 mL/L)后所测得的荧光强度值;Fmin为最小荧光强度值,即在Fmax基础上加入EGTA(终浓度为5 nmol/L)后所测得的荧光强度值。[Ca2+]i浓度计算:[Ca2+]i=Kd(F-Fmin)/(Fmax-F)(nmol/L),其中Kd=224 nmol/L,为Fura-2AM与Ca2+的解离常数[6]。
1.7 Western blot检测蛋白表达 各组大鼠于预定时间点灌注取脑,取部分顶叶皮层,用细胞膜蛋白试剂盒提取细胞膜蛋白,以BSA为标准,用BCA法进行蛋白定量。取30 μg蛋白样品,SDS-PAGE 电泳,半干法转移至PVDF膜,将膜放入50 g/L脱脂牛奶中37 ℃封闭2 h后,加入一抗(TRPC1,1∶1 000;NeuN,1∶500;β-actin,1∶1 000)4 ℃孵育过夜。TBST缓冲液洗膜后,将膜与辣根过氧化物酶标记的二抗(1∶5 000)室温孵育1 h,洗膜后,用增强化学发光法观察显影,凝胶成像系统拍照后采用ImageJ软件测定条带吸光度作定量分析。
1.8 TUNEL检测皮层神经元凋亡 DAI模型组、干预模型组及正常组各3只大鼠在预定的时间点深度麻醉后,经左心室灌注固定后迅速开颅取脑,40 g/L多聚甲醛溶液中固定24 h,石蜡包埋后连续冠状切片,每个脑块10张,片厚5 μm。按照TUNEL凋亡试剂盒提供步骤进行TUNEL染色。图像分析系统采集图像,每张切片随机选取5个高倍视野(×400),计数TUNEL阳性神经元[7]。
2 结 果
2.1 皮层神经元及轴索的形态学变化 HE染色可见DAI模型组皮层下组织疏松,神经元形态异常,有空泡、核碎裂、核固缩等形成,在靠近大脑中线附近皮层中可见点状出血,部分大脑皮层下可见小神经胶质细胞增生及微血管生成,而在正常组中皮层下组织结构紧密,神经元形态正常,胶质细胞分布均匀(图1A~D)。NeuN染色可见正常组中神经轴突形态正常,树突结构连续,树突形态光滑,DAI模型组中可见轴缩球形成,树突部分形成串珠状结构(图1E、F)。透射电镜下可见DAI模型组中神经元肿胀,其内线粒体、高尔基体、内质网扩张,嵴紊乱,周围有高密度电子物质生成,细胞核变形,胞质内空泡形成,染色质不均匀、边集、浓缩;正常组中可见髓鞘形态完整,结构连续,其内可见神经纤维断层;DAI模型组中可见髓鞘正常结构被破坏,轴突断裂,髓鞘肿胀,髓鞘连续性片层结构疏松、分离或消失,片层结构内缺乏神经纤维和线粒体(图1G~J)。
2.2 [Ca2+]i变化 DAI模型组中皮层神经元[Ca2+]i在1 d时明显升高,之后逐渐降低,至10 d时降至最低。SKF96365干预模型组中皮层神经元[Ca2+]i虽在1 d开始上升,总体升高趋势被明显抑制,高于正常组,与DAI模型组相比较有统计学意义(图2,P<0.05)。
2.3 TRPC1在皮层中表达变化 免疫组化染色显示,DAI后TRPC1可在大脑皮层中广泛表达,并主要在大脑板层Ⅱ至V中表达;其中DAI后1 d表达显著,之后表达逐渐降低,10 d时表达已较弱(图3A~E)。SKF96365干预模型组中TRPC1在1 d时表达可被抑制,其表达趋势未出现显著增加(图3F~J)。Western blot结果显示,DAI模型组皮层中TRPC1蛋白的表达在1 d时最高,之后呈逐渐下降趋势,至10 d达到最低,灰度值分析显示DAI模型组1 d及3 d与其他各组相比有统计学意义(图4A,P<0.05);NeuN在DAI后表达明显降低,在1 d时最显著,与其他时间段相比有统计学意义,之后逐渐升高(图4A,P<0.05)。SKF96365干预模型组中TRPC1蛋白的表达明显被抑制,灰度分析显示各个时间段之间相比无统计学意义(图4B,P>0.05);NeuN在干预模型组中表达呈现逐渐增高趋势,在5 d 时达到峰值,之后逐渐降低(图4B)。
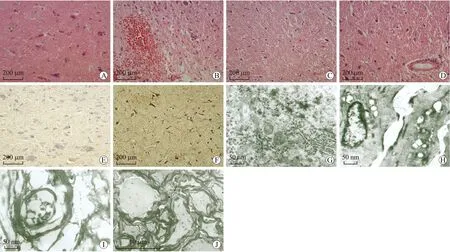

图1 皮层神经元及轴索形态学的变化Fig.1Morphologicalchangesofcorticalneuronsandaxon
A:正常大鼠皮层结构(HE, ×200);B:DAI模型组1 d的皮层结构,见皮层下有出血点;C:DAI模型组3 d皮层结构疏松;D:DAI模型组5 d组皮层下有血管生成;E:正常组NeuN染色,神经元轴突及树突结构正常(×100);F:DAI模型组NeuN染色,神经元轴突存在,树突不连续,有轴缩球形成(×100);G:DAI模型组神经元内线粒体、内质网及高尔基体扩张,可见高密度电子物质(×10 000);H:DAI模型组中肿胀的神经元及周围空泡形成(×10 000);I:DAI模型组中髓鞘变性,正常结构破坏,神经纤维消失(×10 000);J:SKF96365干预组中髓鞘结构部分保留,其内神经微丝存在(×4 000)。
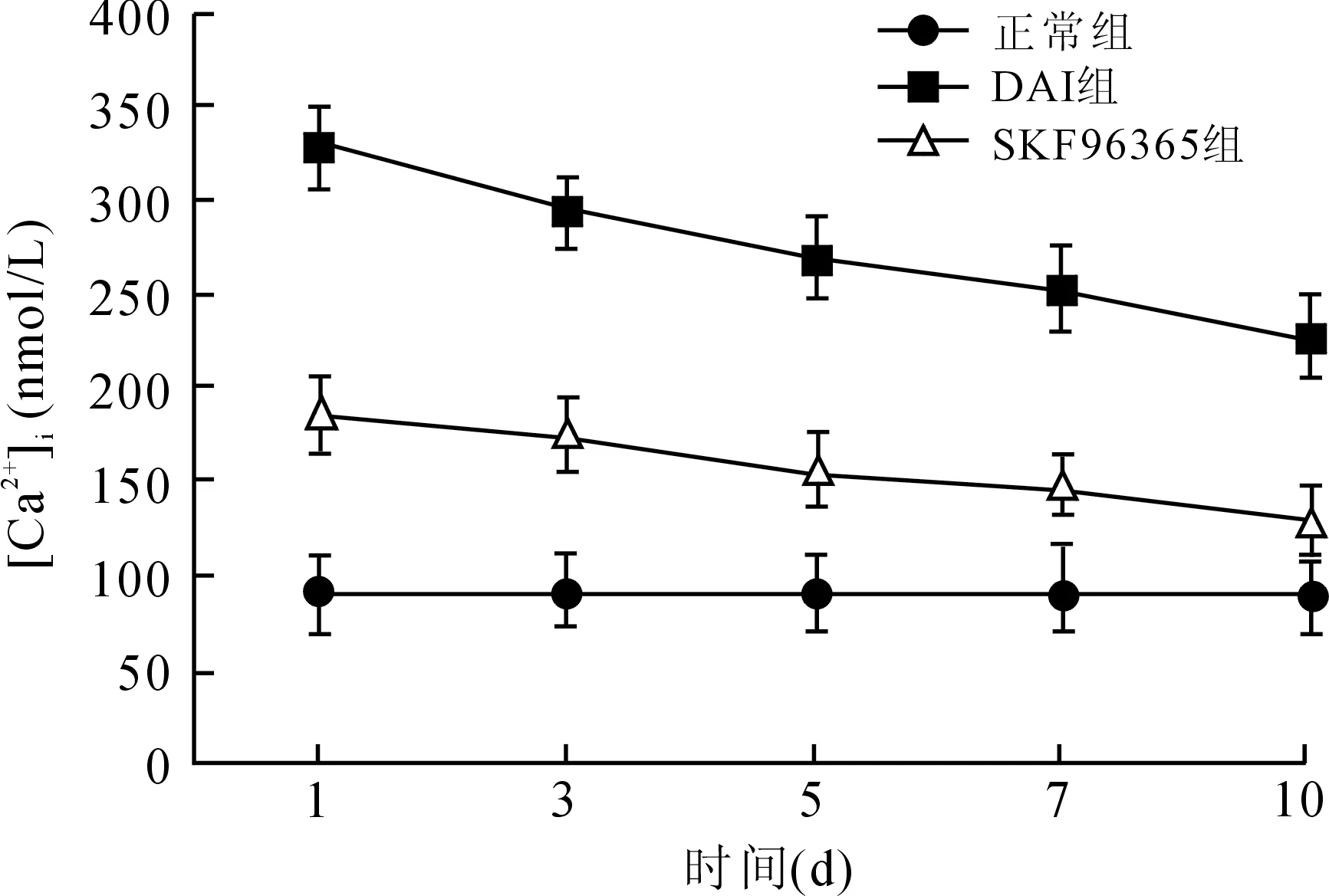
图2 DAI模型组及SKF96365干预组[Ca2+]i变化
Fig.2 Changes of intracellular calcium concentration in DAI group and SKF96365 intervention DAI group
2.4 TUNEL阳性神经元表达及计数 TUNEL染色结果显示,DAI模型组在1 d时已有皮层神经元的TUNEL阳性细胞,之后逐渐增多,至5 d时最多;SKF96365干预模型组中TUNEL阳性细胞虽较正常组有增高,但其显著增高趋势被抑制。TUNEL阳性细胞计数表明,DAI模型组中3、5、7 d与SKF96365干预模型组对比有统计学意义(图5、图6,P<0.05)。
2.5 大鼠神经功能缺损评分 DAI模型组动物在建模后均出现精神淡漠、嗜睡、活动减少、饮水和进食减少,体质量较术前明显减轻。而SKF96365干预模型组以上大体表现在1 d后出现明显改善。DAI模型组各个时间段神经功能评分与SKF96365干预模型组各个时间段神经功能评分相比差异有统计学意义(表1,P<0.05)。
表1 大鼠神经功能缺损评分结果

Tab.1 The neurological severity scores of rats in different groups (±s)
与正常对照组比较,*P<0.05,与DAI模型组比较,△P<0.05。
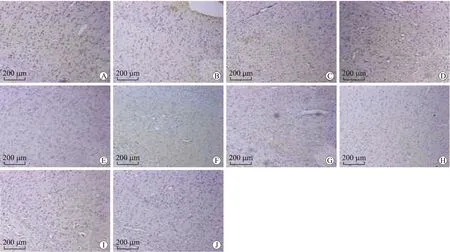

图3 DAI模型组及SKF96365干预模型组TRPC1在大脑皮层的免疫组化染色Fig.3ImmunohistochemicalstainingofTRPC1inthecere-bralcortexofDAIgroupandSKF96365interventiongroup
DAI模型组:A:1 d组皮层TRPC1免疫组化染色,此时表达最明显;B:3 d组皮层TRPC1免疫组化染色,表达较前下降;C:5 d组皮层TRPC1免疫组化染色,表达进一步下降;D:7 d组皮层TRPC1免疫组化染色,表达明显下降;E:10 d组皮层TRPC1免疫组化染色,表达接近正常。SKF96365干预模型组:F:1 d组皮层TRPC1免疫组化染色;G:3 d组皮层TRPC1免疫组化染色;H:5 d组皮层TRPC1免疫组化染色;I:7 d组皮层TRPC1免疫组化染色;J:10 d组皮层TRPC1免疫组化染色。F~J各组表达未出现明显改变。

图4 DAI模型组及SKF96365干预模型组皮层TRPC1及NeuN蛋白表达及灰度值分析
Fig.4 The protein expressions and gray value analysis of TRPC1 and NeuN in DAI group and SKF96365 intervention group
A:DAI模型组1、3 d时TRPC1蛋白表达明显升高,与其他各组相比有统计学意义(*P<0.05);NeuN表达在1 d时减少,之后逐渐升高。B:DAI干预模型组[Ca2+]iTRPC1蛋白表达升高不显著;NeuN表达在3 d时升高,5 d达峰值,之后有所下降。
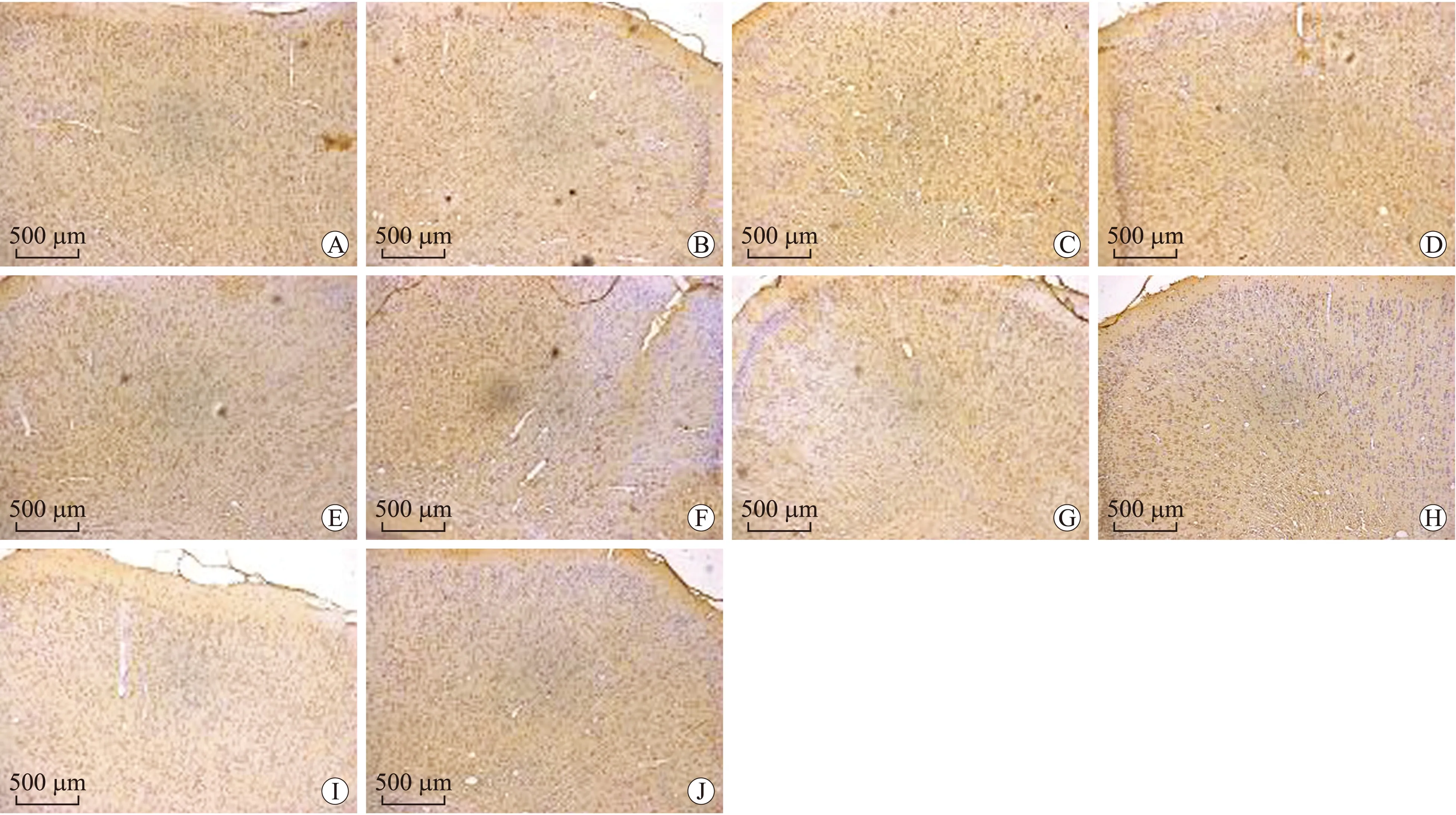

图5 DAI模型组及SKF96365干预模型组皮层TUNEL染色Fig.5TUNELstainingofTUNEL-positivecellsinDAIgroupandSKF96365interventiongroup
DAI模型组:A:1 d组皮层TUNEL染色;B:3 d组皮层TUNEL染色,TUNEL阳性细胞表达增多;C:5 d组皮层TUNEL染色,TUNEL阳性细胞表达最多;D:7 d组皮层TUNEL染色,TUNEL阳性细胞表达开始减少;E:10 d组皮层TUNEL染色,TUNEL阳性细胞显著减少。SKF96365干预模型组:F:1 d组皮层TUNEL染色;G:3 d组皮层TUNEL染色;H:5d组皮层TUNEL染色;I:7 d组皮层TUNEL染色;J:10 d组皮层TUNEL染色。F~J:TUNEL阳性细胞表达被显著抑制。
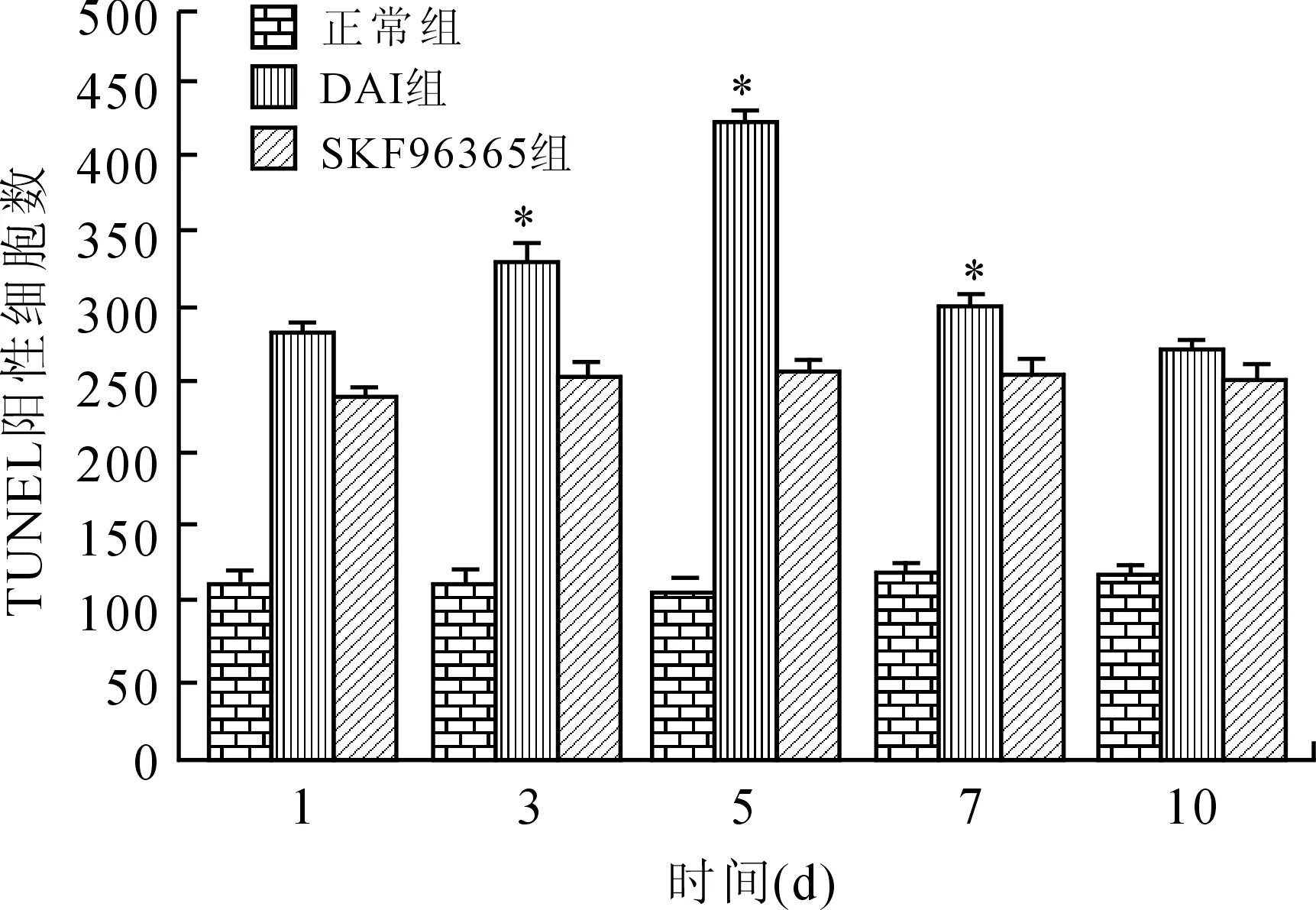
图6 DAI模型组及SKF96365干预模型组皮层TUNEL阳性细胞计数
Fig.6 Cell count of TUNEL-positive cells in DAI group and SKF96365 intervention group
与同时间点SKF96365组相比,*P<0.05。
3 讨 论
TBI后病理生理机制的研究有赖于可靠的动物及细胞模型的建立。目前,有关TBI动物模型建立的方法主要包括有液压冲击模型、控制性皮层损伤模型、自由落体模型及瞬间旋转损伤模型等[8],而目前仅有瞬间旋转动物模型可较好的模拟DAI后病理生理改变。本研究团队既往利用自制瞬间旋转损伤模型较好模拟了DAI后病理生理改变[4,9-10]。在瞬间旋转损伤后,经HE染色、组织化学染色及电镜检测可发现,DAI后位于大脑中线的皮层下组织结构疏松性改变,可伴有点状出血,有轴缩球形成,树突部分形成串珠状结构,神经元肿胀,细胞器扩张,胞核变形,胞质空泡形成,髓鞘结构被破坏,轴突断裂,片层结构内神经纤维缺乏。这些改变符合DAI后皮层的病理改变,说明本实验所采用的瞬间旋转损伤模型是可靠的。在DAI发生时,大脑白质内所有的轴突均经历了类似的动态损伤过程。在某些特定区域,如大脑中线附近的轴突出现水肿,造成离子转运体异常聚集,导致正常离子转运障碍;即使其周围正常轴突并未出现离子转运障碍,这些轴突仍可发生病理生理学改变,导致轴突功能障碍,如在轻度创伤性脑损伤(mild traumatic brain injury, mTBI)后可观察到神经传导速度的改变[11]。
TRPC1是哺乳动物TRP家中第一个被认为可形成离子通道的成员[12]。在初期研究中,TRPC1的表达可使由Ca2+库耗竭激活的非选择性阳离子电流升高。由于其广泛表达,TRPC1的功能与其他TRPC亚基可在体外[13]和体内[14]共同作用。本研究选择TRPC1作为目标蛋白,研究其在DAI后的作用及可能的机制。SKF96365可抑制TRPC离子通道的活性,减少钙内流,是TRPC离子通道常用阻断剂之一[15],本研究通过立体定向侧脑室显微注射将SKF96365注入脑脊液循环中。本实验结果表明,DAI后TRPC1的表达增高,在1d时最为明显,此后逐渐下降,而针对皮层神经元[Ca2+]i的检测发现,DAI后[Ca2+]i的变化趋势与TRPC1变化趋势相一致,在建模后1d时[Ca2+]i最高,之后逐渐降低。应用TRPC1通道阻断剂干扰其活性后,TRPC1表达增高趋势被明显抑制,而[Ca2+]i的升高也被抑制,说明TRPC1与神经元胞内钙内流趋势呈正相关。前期研究已证实[2],在胞外Ca2+缺失的情况下,大鼠视神经及脊髓后索神经中轴质Ca2+水平的增加是为了应对体外有髓鞘轴突的缺血,这表明胞内钙库在轴突变性中起重要作用。胞外Ca2+通过轴膜内流,在缺血性脊髓后索轴突中,轴突内钙超载通过兰尼碱受体介导的内质网钙内流而发生,还可通过应对磷脂酶C(PLC)介导IP3生成的1,4,5,-磷酸腺苷受体来释放,或可通过线粒体中的Ca2+释放。轴索损伤后的离子失衡被认为在轴索损伤后轴突变性和其他未受损轴突的持续功能障碍中起关键作用。近来有研究利用轴突牵张损伤模型发现,[Ca2+]i的急性升高可能部分来自于胞内钙库的释放[16]。
研究表明,脱髓鞘在TBI的病理生理过程中亦发挥重要作用。本研究中,利用电镜观察到DAI后正常髓鞘结构发生明显改变,轴突断裂,髓鞘肿胀,髓鞘连续性片层结构疏松、分离或消失,片层结构内缺乏神经纤维和线粒体。而抑制TRPC1介导的钙内流后,DAI后髓鞘正常结构可得到部分保存,大鼠神经功能缺损评分亦得到明显改善。有研究发现,豚鼠视神经的牵张性损伤后可发生髓鞘的急性断裂。但是,尚不清楚髓鞘损伤的发生是否仅为轴突变性的直接结果。临床上可在急性或慢性TBI后观察到凋亡性少突神经胶质细胞,而少突神经胶质细胞的丢失可能造成髓鞘形成不足,潜在的影响轴突完整性及其功能[17]。
我们在前期研究中发现胞内Ca2+感受器STIM1可在DAI后早期脑损伤中发挥重要作用[9],而TRPC1通道被认为亦可发挥类似于钙库操控性钙通道(store-operated calcium channels, SOCs)的功能,作为SOCC通道的筛选亚型而引起关注,研究主要集中在TRPC通道的受体激活和库依赖性钙内流方面[18]。近期研究表明,STIM1调控TRPC通道的开放,TRPCs与Orais可共同作用。虽然许多研究支持TRPCs可发挥SOCs的作用,但其作用似乎仅存在于部分细胞系或特定条件下。初期研究表明,STIM1与多种TRPC通道结合,并门控TRPCs[19]。随后许多研究证实了STIM1和TRPC1之间的相互作用[20]。然而,近期有学者发现未有证据证明TRPC1钙库依赖调控的,但报道了其在Ca2+游离溶液中可被DAG激活。研究发现,在生理状态下当细胞被激活PLC-β的激动剂所刺激时,TRPC1的表达并不造成可检测到的离子电流[14]。TRPC1可能是不同异源性TRP复合物的一个组分,其是否可在其他TRP亚基不存在的情况下形成功能性通道目前尚未有研究。
综上所述,DAI后早期皮层高表达的TRPC1通道蛋白,是造成髓鞘变性及神经元钙内流的重要原因,过度钙信号的传入可触发下游细胞凋亡机制,引起脑损伤;抑制TRPC1的表达可减少神经元凋亡,抑制髓鞘变性,改善神经功能。而TRPC1与STIM1是否在DAI后共同发挥SOCC作用,尚需进一步研究证实。
[1] POVLISHOCK JT, KATZ DI. Update of neuropathology and neurological recovery after traumatic brain injury[J]. J Head Trauma Rehabil, 2005, 20(1):76-94.
[2] JPNIKOLAEVA MA, MUKHERJEE B, STYS PK. Na+-dependent sources of intra-axonal Ca2+release in rat optic nerve duringinvitrochemical ischemia[J]. J Neurosci, 2005, 25(43):9960-9967.
[3] OUARDOUZ M, NIKOLAEVA MA, CODERRE E, et al. Depolarization-induced Ca2+release in ischemic spinal cord white matter involves L-type Ca2+channel activation of ryanodine receptors[J]. Neuron, 2003, 40(1):53-63.
[4] 刘晓斌,宋锦宁,陈景宇,等. 脑弥漫性轴索损伤实验装置的研制及动物模型的建立[J]. 西安交通大学学报:医学版, 2008, 29(5):595-598.
[5] CHEN J, SANBERG PR, LI Y, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats[J]. Stroke, 2001, 32(11):2682-2688.
[6] DILDY JE, LESLIE SW. Ethanol inhibits NMDA-induced increases in free intracellular Ca2+in dissociated brain cells[J]. Brain Res, 1989, 499(2):383-387.
[7] CARLONI S, CARNEVALI A, CIMINO M, et al. Extended role of necrotic cell death after hypoxia-ischemia-induced neurodegeneration in the neonatal rat[J]. Neurobiol Dis, 2007, 27(3):354-361.
[8] O’CONNOR WT, SMYTH A, GILCHRIST MD. Animal models of traumatic brain injury: a critical evaluation[J]. Pharmacol Ther, 2011, 130(2):106-113.
[9] LI Y, SONG J, LIU X, et al. High expression of STIM1 in the early stages of diffuse axonal injury[J]. Brain Res, 2013, 1495:95-102.
[10] SONG JN, LIU XB, CHEN J, et al. Dynamic changes in cerebral microcirculation and hypoxia in the early stages of diffuse axonal injury. [J]. Neural Regeneration Res, 2011, 6(20):1530-1536.
[11] REEVES TM, PHILLIPS LL, LEE NN, et al. Preferential neuroprotective effect of tacrolimus (FK506) on unmyelinated axons following traumatic brain injury[J]. Brain Res, 2007, 1154:225-236.
[12] ZITT C, ZOBEL A, OBUKHOV AG, et al. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion[J]. Neuron, 1996, 16(6):1189-1196.
[13] LINTSCHINGER B, BALZER-GELDSETZER M, BASKARAN T, et al. Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels[J]. J Biol Chem, 2000, 275(36):27799-27805.
[14] STRUBING C, KRAPIVINSKY G, KRAPIVINSKY L, et al. TRPC1 and TRPC5 form a novel cation channel in mammalian brain[J]. Neuron, 2001, 29(3):645-655.
[15] RAE MG, HILTON J, SHARKEY J. Putative TRP channel antagonists, SKF 96365, flufenamic acid and 2-APB, are non-competitive antagonists at recombinant human alpha1beta2gamma2 GABA(A) receptors[J]. Neurochem Int, 2012, 60(6):543-554.
[16] VON REYN CR, SPAETHLING JM, MESFIN MN, et al. Calpain mediates proteolysis of the voltage-gated sodium channel alpha-subunit[J]. J Neurosci, 2009, 29(33):10350-10356.
[17] SHAW K, MACKINNON MA, RAGHUPATHI R, et al. TUNEL-positive staining in white and grey matter after fatal head injury in man[J]. Clin Neuropathol, 2001, 20(3):106-112.
[18] KISELYOV K, KIM JY, ZENG W, et al. Protein-protein interaction and functionTRPC channels[J]. Pflugers Arch, 2005, 451(1):116-124.
[19] HUANG GN, ZENG W, KIM JY, et al. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels[J]. Nat Cell Biol, 2006, 8(9):1003-1010.
[20] ZENG W, YUAN JP, KIM MS, et al. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction[J]. Mol Cell, 2008, 32(3):439-448.
(编辑 韩维栋)
The mechanism of high expression of TRPC1 in diffuse axnoal injury of rats
ZHANG Ming1,2, SONG Jin-ning2, LI Yu2, ZHAO Yong-lin2,LI Dan-dong2, WU Yuan3, FU Zhou-feng2, ZHANG Bin-fei2
(1. Department of Neurosurgery, the Second Affiliated Hospital, Medical School ofXi’an Jiaotong University, Xi’an 710004; 2. Department of Neurosurgery,the First Affiliated Hospital, Medical School of Xi’an Jiaotong University, Xi’an 710061;3. Department of Critical Care Medicine, the Second Affiliated Hospital,Medical School of Xi’an Jiaotong University, Xi’an 710004, China)
Objective To investigate the possible role of canonical transient receptor potential channel 1 (TRPC1) after diffuse axonal injury (DAI) and discuss the mechanism of TRPC1 involved in calcium overload of neurons and myelin degeneration after DAI. Methods Rat models of DAI were established with the method of rotational acceleration of the brain. DAI intervention group was established by using SKF96365 intracerebroventricular injection. Intracellular calcium concentration was detected by Fura-2AM. The dynamic expressions of TRPC1 in the cortex were determined by Western blot and immunohistochemical staining. The results were compared with those in DMSO control group and normal group. The changes of neurons and myelin sheath were detected under electron microscope. The apoptosis of cortical neurons was detected by TUNEL. One-way ANOVA was used to compare within each group. Results The protein expressions of TRPC1 in the cortex increased after DAI and reached the peak at day 1 and then gradually decreased while intracellular calcium concentration also reached the peak at day 1. The structure of myelin sheath was destructed at the same time. After injection of TRPC1 blocker SKF96365, the protein expression of TRPC1 was inhibited and intracellular calcium concentration decreased. Meanwhile, the structure of myelin sheath was partially reserved and the scores of neurological functions in rats were improved. Conclusion TRPC1-induced calcium overload after DAI is the major cause of myelin degeneration and neuronal death. Suppressing the calcium influx induced by TRPC1 can protect the normal structure of myelin and improve neuronal apoptosis.
transient receptor potential channel 1 (TRPC1); diffuse axonal injury(DAI); myelin degeneration; calcium overload; neuronal apoptosis
2014-04-21
2014-06-26
国家自然科学基金资助项目(No.30471774);教育部新世纪优秀人才支持计划资助项目(No.NCET-05-0831);陕西省自然科学基金资助项目(No.2003C1-16) Supported by the National Natural Science Foundation of China (No.30471774),the New-Century Excellent Talents Program of Ministry of Education (No.NCET-05-0831) and the Natural Science Foundation of Shaanxi Province (No.2003C1-16)
宋锦宁,博士,教授,主任医师,博士生导师. E-mail: jinnings@126.com
张明(1981-),男(汉族),博士,主治医师. 研究方向:脑血管病、颅脑损伤的基础与临床. E-mail: phoenixzhang@126.com
时间:2014-09-19 16∶34 网络出版地址:http://www.cnki.net/kcms/detail/61.1399.R.20140919.1634.005.html
R651.1
A
10.7652/jdyxb201406004
猜你喜欢
中华耳科学杂志(2022年1期)2022-11-24
生物化学与生物物理进展(2022年11期)2022-03-02
浙江临床医学(2021年12期)2021-11-29
健康之家(2021年19期)2021-05-23
世界最新医学信息文摘(2020年25期)2020-12-25
党的生活(黑龙江)(2018年9期)2018-10-17
益寿宝典(2018年1期)2018-01-27
中国卫生标准管理(2015年4期)2016-01-14
中华神经创伤外科电子杂志(2015年1期)2015-01-21
西南医科大学学报(2014年6期)2014-03-20
