
ω -芋螺毒素MVIIA 和MVIIC 对α 9 α 10乙酰胆碱受体的活性研究
2014-07-10 12:21长孙东亭朱晓鹏胡远艳罗素兰
海南大学学报(自然科学版) 2014年4期
长孙东亭,朱晓鹏,吴 勇,胡远艳,罗素兰
(海南大学热带生物资源教育部重点实验室 海口市海洋药物重点实验室,海南 海口570228)
来自热带海洋的肉食性软体动物—芋螺毒液中的各种毒素肽,被称为芋螺毒素或芋螺肽(Conotoxin,Conopeptide,CTx),它们是富含二硫键的小肽,大多由10 ~40 个氨基酸残基组成,其分子量往往小于5 kDa,具有特异阻断动物体内各种膜受体、离子通道、转运蛋白等的特殊功能,具有巨大的药物开发潜力[1].芋螺毒素种类繁多,根据其前体蛋白内质网信号肽序列的相似性以及其半胱氨酸模式,可将其分为不同的基因超家族,至今,所有已知的芋螺毒素被分为19 个超家族,分别为A,B,C,D,S,M,I1,I2,I3,J,L,O1,O2,O3,P,T,V,Y,K[2-4].根据芋螺毒素作用受体的靶位,可将其分为ω、μ、δ 等多种药理学家族.根据每个超家族的受体靶标类型,还可将其分为A,κA(A -超家族);ω,δ,κ,μO(O -超家族);μ,ψ,κM(M -超家族)等家族(亚型).其中ω-芋螺毒素是作用于钙离子通道的,其半胱氨酸模式为C—C—CC—C—C,含有3 对二硫键,属于O-超家族,一般由25 ~35 个氨基酸残基组成.ω-芋螺毒素是作用于乙酰胆碱受体(nAChRs)的,其半胱氨酸模式为CC—C—C,含有2 对二硫键,一般由13 ~20 个氨基酸残基组成[3].
研究表明,ω - 芋螺毒素特异阻断的N - 型钙离子通道(Cav2. 2)[5]和α9 α10 乙酰胆碱受体(nAChR)[6]都是不成瘾的、治疗神经痛的新型镇痛药物的作用靶点,前者通过中枢神经系统(CNS)给药(如鞘内给药),后者则通过外周神经系统(CNS)给药(如肌肉注射).在世界范围内,无论是在临床上,还是在社会上和经济上,神经痛(慢性疼痛、顽固性疼痛)都是一个难题,这种疼痛折磨着地球上8%以上的人口[7-9],消耗了大量的医疗资源,而且至今尚不能得到很好的治疗.反复发作的慢性疼痛与其复杂的病理或生理机制密切相关.改变感觉神经元中N-型钙离子通道(N-type VGCC)[10-13]或者外周神经系统中的α9 α10 乙酰胆碱受体(nAChR)的功能,是涉及这些复杂疼痛机制的一个重要方面,可望达到治疗慢性疼痛的目的[14-16].
已知ω-芋螺毒素MVIIA 是N-型钙离子通道(Cav2.2)的特异阻断剂(见图1A,表1),人工合成的MVIIA 多肽(Ziconotide,奇考诺肽)已被美国FDA 和欧洲药品管理局批准为治疗新药,用于治疗严重的慢性疼痛[17].
ω-芋螺毒素MVIIC 是N -型(Cav2.2)和P/Q -型钙离子通道(Cav2.1)的特异阻断剂(见图1B,表1)[18-19].因而,N-型钙离子通道被认为是镇痛药的作用靶点,在疼痛信号的加工过程中起着很重要的作用.
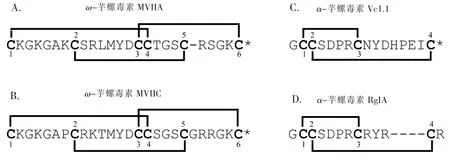
图1 ω-芋螺毒素MVIIA 和MVIIC 与α-芋螺毒素Vc1.1 和RgIA 的序列及二硫键的连接方式(图中*表示C 末端酰胺化)

表1 ω -芋螺毒素MVIIA 和MVIIC 与α -芋螺毒素Vc1.1 和RgIA 的靶点活性比较
已知α-芋螺毒素Vc1.1 和RgIA 原本是神经型α9 α10 乙酰胆碱受体(nAChR)的特异阻断剂(见图1C─D 和表1),在神经痛模型上,通过肌肉注射具有急性和持久性的镇痛作用[16],令人吃惊的是,α -芋螺毒素Vc1.1 和RgIA 也能阻断经由G -蛋白偶联的GABAB 受体介导的N -型钙离子通道(见图1C─D,表1)[20].那么,反过来,ω-芋螺毒素能否阻断α9 α10nAChR 受体呢?为此,本文针对2 个ω-芋螺毒素MVIIA 和MVIIC,研究了它们对α9 α10 nAChR 的有效阻断,旨在阐明不同或同一芋螺毒素与α9 α10 nAChR 和钙离子通道之间的关系,这对于研发新型的芋螺毒素镇痛药和更好地治疗顽固性疼痛具有很重要的科学意义.
1 材料与方法
1.1 乙酰胆碱受体(nAChRs)α9 和α10 亚基cRNA 的制备 大鼠神经型乙酰胆碱受体的α9 和α10 亚基克隆来自A.B. Elgoyhen (Buenos Aires,Argentina).用中量质粒抽提试剂盒分别提取含α9 和α10 亚基基因的质粒,先用限制性内切酶对质粒进行酶切线性化,然后以纯化后的线性DNA 作为体外转录的模板.用mMESSAGE mMACHINE Kit 体外转录试剂盒(Ambion ),分别合成α9 和α10 亚基的cRNA,进一步用RNA 纯化试剂盒纯化后,在260 nm 波长下测定并计算cRNA 的浓度,分装后于-70 ℃保存备用.
1.2 非洲爪蟾卵母细胞的分离和培养 参照文献[21]中的优化方法,选取成熟的美国进口的雌性非洲爪蟾(Xenopus laevis)(见图2),将其埋在碎冰中,冰浴麻醉45 min 左右,待其完全麻痹后,仰卧状放置在冰面上,按手术规程取出足量的卵叶,剪碎后浸泡于盛有OR2 溶液(41 mmol·L-1NaCl,1 mmol·L-1KCl,0.5 mmol·L-1MgCl2,2.5 mmol·L-1HEPES,pH 7.1 ~7.7)的培养皿中,用OR2 溶液将卵母细胞清洗干净,在含有0.5 g·L-1胶原酶的OR2 溶液中于室温下振荡消化约1 h,胶原酶消化2 次,然后再用OR2 溶液清洗干净,挑选分离良好、健康而有弹性的卵母细胞(见图2),转移到含抗生素的普通ND96 培养液中,于17℃恒温培养箱中培养.普通ND96 培养液的组成为:96 mmol·L-1NaCl,2 mmol·L-1KCl,1 mmol·L-1CaCl2,1 mmol·L-1MgCl2,5 mmol·L-1HEPES,pH 7.4.卵母细胞培养是在ND96 中,另加3 种抗生素,即青霉素10 mg·L-1,链霉素10 mg·L-1,庆大霉素100 mg·L-1.

图2 用于表达α9α10 nAChR 的非洲爪蟾(左)和分离良好的卵母细胞(右)
1.3 α9 α10 nAChR 的表达及其电生理记录 将新鲜分离的非洲爪蟾卵母细胞培养过夜后,根据α9 和α10 亚基cRNA 的浓度,按照终浓度比例为1∶ 1 的原则,将α9 和α10 的cRNA 混合均匀后,注入卵母细胞中,每个亚基的注射量为:每个细胞15 ng cRNA.注射后置于ND96 培养液中,于17 ℃下培养.注射后第2 ~5 天,利用双电极电压钳系统检测α9 α10 nAChR 的电流表达情况,并进行电生理记录,具体过程简述如下:将1 个注射过α9 和α10 cRNA 的蛙卵置于30 μL 的Sylgard 记录槽中,重力灌注含有0.1 g/L BSA(bovine serum albumin)的不含抗生素的ND96 灌流液,流速为每分钟1 ~2 mL(加入BSA 的目的是减少毒素的非特异性吸附).用自动转换进样器在配体开放剂乙酰胆碱(Ach)和ND96 灌流液之间进行切换.Ach门控的电流由双电极电压钳放大器设置在“慢”钳,并于“clamp gain”在最大值(×2 000)位置时进行在线记录.用1 mm 外径×0.75 mm 内径的玻璃毛细管(fiber-filled borosilicate capillaries,WPI Inc.,Sarasota,FL)拉制玻璃电极,并以KCl(3 mmol·L-1)作为电压和电流电极,膜电压钳控制在-70 mV,整个系统均由电脑控制和记录数据.神经型α9 α10 nAChR 的ACh 浓度为10 μmol·L-1,ACh 脉冲为每隔1 分钟自动灌注1 秒的ACh,并采用电脑记录每个ACh 脉冲产生的电流大小和轨迹.
1.4 ω -芋螺毒素MVIIA 和MVIIC 对α9 α10 nAChR 的结合活性测定 ω-芋螺毒素MVIIA 和MVIIC(见图1A―B)以及阳性对照α-芋螺毒素RgIA(见图1D)由美国犹他大学的J M McIntosh 教授提供.本研究分别测定了它们在普通ND96 灌流液和不含钙离子的钡离子ND96(Ba2+-ND96)灌流液中对α9 α10 nAChR 的结合活性.普通ND96 灌流液是含有0.1 g·L-1BSA 的普通ND96 培养液;钡离子ND96 灌流液是将普通ND96 灌流液中的CaCl2用等摩尔浓度的BaCl2来代替. 将ω -芋螺毒素MVIIA 和MVIIC以及阳性对照α-芋螺毒素RgIA 分别用含有0.1 g·L-1BSA 的普通ND96 灌流液和钡离子ND96 灌流液配制成100 μmol·L-1的高浓度溶液,然后再用相同的灌流液稀释至10 μmol·L-1,电流记录时,按1/10 体积(3 μL)的比例加入到记录槽中,毒素终浓度分别为10 μmol/L 和1 μmol·L-1.在测定毒素对α9 α10 nAChR 的结合活性时,ACh 脉冲为:每隔5 min 自动灌注1 s 的ACh,在产生毒素作用的第一个电流轨迹之后,连续用每隔1 分钟自动灌注1 秒 的ACh 脉冲,一直用灌流液对毒素进行冲洗,并观察毒素的洗脱速度.用与毒素溶液相等体积的ND96 灌流液或钡离子ND96 灌流液(3 μL)作为相应的对照电流,以对照电流为分母,并以不同毒素浓度的电流大小为分子,计算电流反应百分数(%).每个毒素浓度的电流反应检测4 ~6 个卵母细胞.
2 结果与分析
2.1 ω -芋螺毒素MVIIA 在普通ND96 中对α9 α10nAChR 的活性 神经型α9 α10 nAChR 在非洲爪蟾卵母细胞中的表达一般需要30 h 以上,注射α9 和α10 cRNA 后的第2 ~5 天均可进行电生理记录.在普通ND96 灌流液中,α9 α10 nAChR 电流中90%以上是由钙离子激活产生的氯离子(Cl-)电流组成的,其电流很大,10 μmol·L-1的ACh 配体浓度所激发的α9 α10 nAChR 电流一般都在3 000 ~20 000 nA 之间,随着表达时间的延长,α9 α10 nAChR 的电流越来越大,有的甚至可达到100 000 nA,太大的电流往往使得膜电压很难钳制在-70 mV,从而影响结果. 因而,表达超过5 d 的爪蟾卵母细胞不再适合做电生理记录.10 μmol·L-1的ω-芋螺毒素MVIIA 在普通ND96 中,对α9 α10 nAChR 显示出极其微弱的活性,图3 显示了其具有代表性的电流轨迹.在图3 中,对照电流C 的大小为12 500 nA,在10 μmol·L-1的MVIIA 条件下,ACh 脉冲产生的电流为10 250 nA,其电流反应百分数为82%.用10 μmol·L-1的MVIIA 重复检测了其他4 个卵母细胞的活性,其结果与图3 相似,电流反应百分数在75% ~90%之间.当用稀释成1 μmol·L-1的MVIIA 来检测时,其已完全没有了阻断活性,其电流反应百分数接近100%,与对照电流已没有差异.
2.2 ω -芋螺毒素MVIIC 在普通ND96 中对α9 α10 nAChR 的活性 10 μmol·L-1的ω -芋螺毒素MVIIC在普通ND96 中,对α9 α10 nAChR 显示出一定的阻断活性,图4A 显示了它的代表性电流轨迹. 在图4A 中,对照电流C 的大小为7 250 nA,在10 μmol·L-1的MVIIC 条件下,ACh 脉冲产生的电流为3 250 nA,其电流反应百分数为45%.用10 μmol·L-1的MVIIC 重复检测了其他5个卵母细胞的活性,其结果与图4A 相似,电流反应百分数在40% ~55%之间,相当于10 μmol·L-1的MVIIC 阻断了α9 α10nAChR 约一半的电流.更换卵母细胞,用稀释成1 μmol·L-1的MVIIC 检测时(见图4B),对照电流C 的大小为10 500 nA,在1 μmol/L 的MVIIC 条件下,ACh 脉冲产生的电流为10 380 nA,其电流反应百分数为99%,几乎没有了阻断活性.对于不同的卵母细胞,1 μmol·L-1的MVIIC 的电流反应百分数都接近100%,与对照电流没有差异.
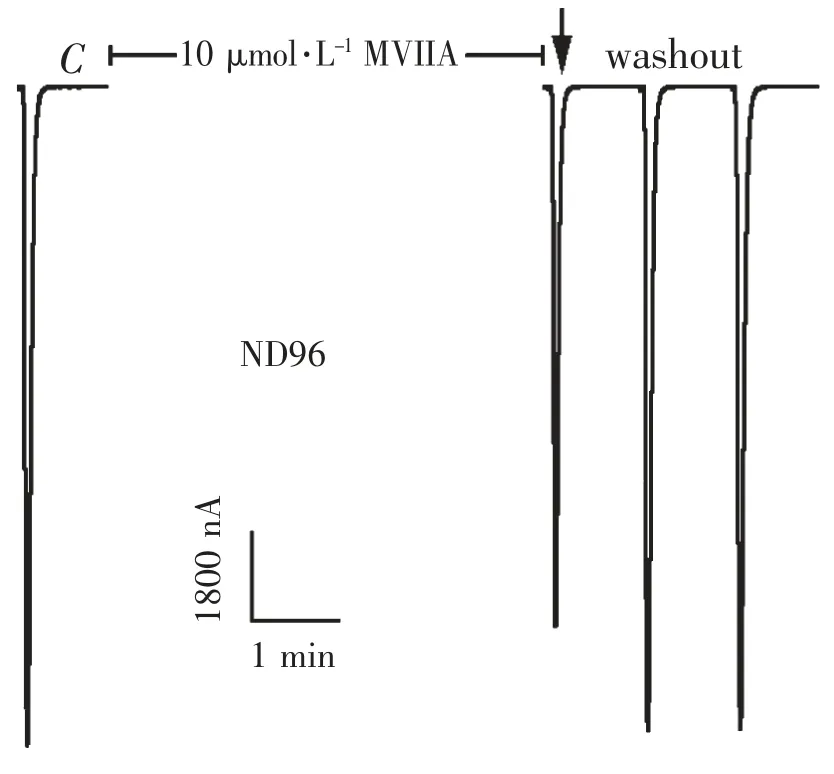
图3 10 μmol·L -1的ω-芋螺毒素MVIIA 在普通ND96 灌流液中对α9 α10 nAChR 的电流影响
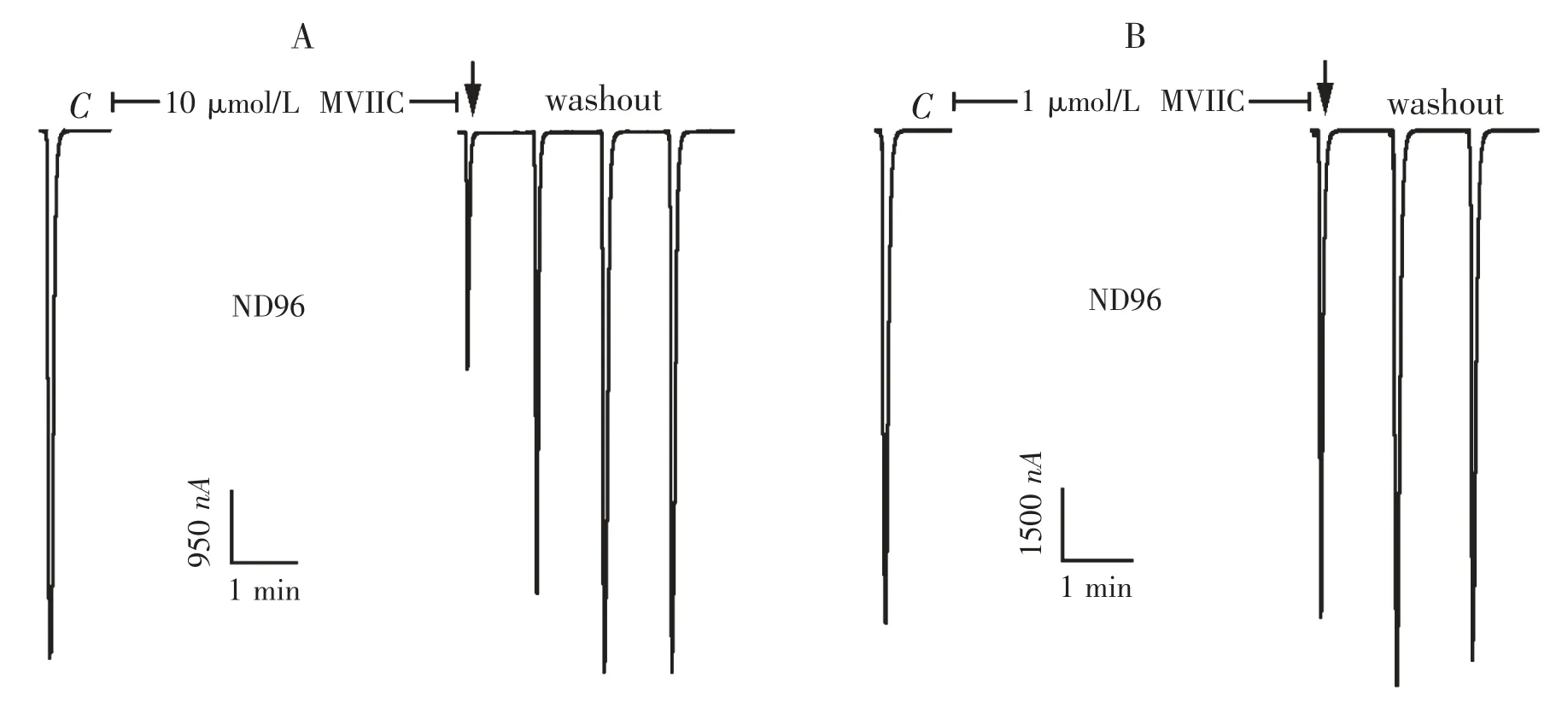
图4 10 μmol·L -1(A)与1 μmol·L -1(B)ω-芋螺毒素MVIIC 在普通ND96 灌流液中对α9 α10 nAChR 的电流影响
2.3 ω -芋螺毒素MVIIA 在钡离子ND96 中对α9 α10nAChR 的活性 α9 α10 nAChR 对钙离子具有很高的通透性,钙离子进入该受体后,会立刻激活氯离子(Cl-)电流,所产生的氯离子电流占据着电生理记录电流的绝大部分.为了排除氯离子电流的影响,本研究用等量的钡离子浓度替换了普通ND96 灌流液中的钙离子浓度,由于没有了钙离子的激活,因此就不会产生氯离子电流了,故电生理记录到的电流全部是α9 α10 nAChR 的真实电流,但该电流较小,不到普通ND96 灌流液电流的10 %,一般在200 ~1 500 nA 之间.在钡离子ND96 灌流液中,可完全排除氯离子电流的干扰[22-23].10 μmol·L-1的ω-芋螺毒素MVIIA在钡离子ND96 中,对α9 α10 nAChR 没有阻断活性,图5 显示了它的代表性电流轨迹.在图5 中,对照电流C 的大小仅为475 nA,在10 μmol·L-1的MVIIA 条件下,ACh 脉冲所产生的电流为490 nA,其电流反应百分数为103%.用10 μmol·L-1的MVIIA 重复检测了其他3 个卵母细胞的活性,其结果都很相似,电流反应百分数在98% ~105%之间,非常接近100%,与对照电流没有差异.
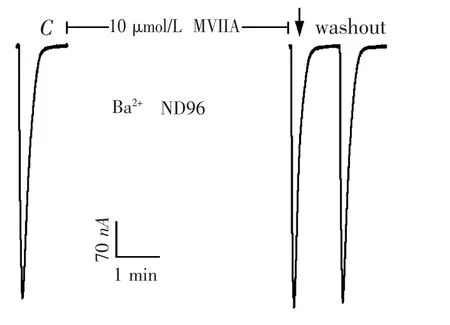
图5 10 μmol·L -1的ω-芋螺毒素MVIIA 在钡离子ND96灌流液中对α9 α10 nAChR 的电流影响
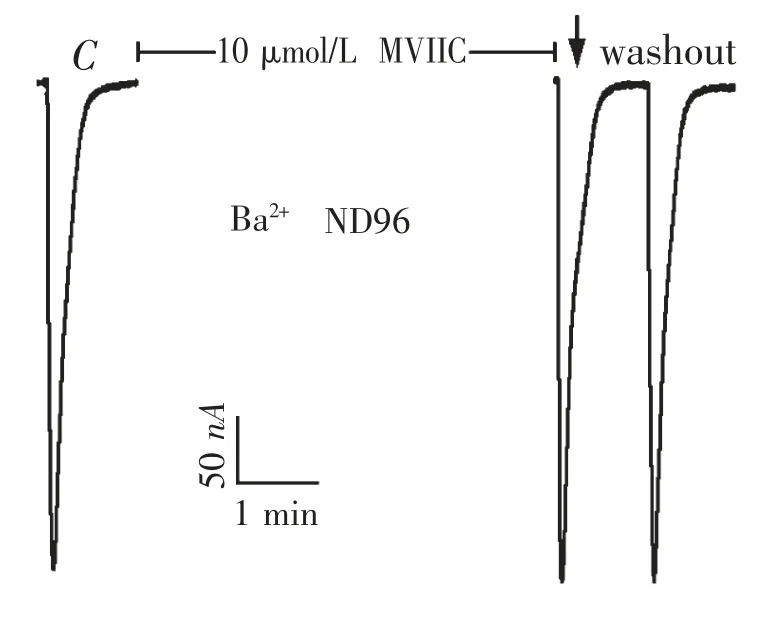
图6 10μmol·L -1 ω-芋螺毒素MVIIC 在钡离子ND96灌流液中对α 9 α 10 nAChR 的电流影响
2.4 ω -芋螺毒素MVIIC 在钡离子ND96 中对α9 α10 nAChR 的活性 10 μmol·L-1的ω-芋螺毒素MVIIC 在钡离子ND96 中对α 9 α 10 nAChR 的活性与10 μmol/L 的MVIIA 的结果类似,也没有阻断活性,图6 显示了其代表性电流的轨迹.在图6 中,对照电流C 的大小为396 nA,在10 μmol·L-1的MVIIC 条件下,ACh 脉冲所产生的电流为403 nA,其电流反应百分数为102%.用10 μmol·L-1的MVIIC 重复检测了其他4 个卵母细胞的活性,其结果都很近似,电流反应百分数在99% ~105%之间,与对照电流完全没有差异.
2.5 α -芋螺毒素RgIA 在普通ND96 和钡离子ND96 灌流液中对α 9 α 10 nAChR 的活性 1 μmol·L-1的阳性对照α-芋螺毒素RgIA 在普通ND96 中,对α 9 α 10 nAChR 显示出极强的阻断活性,图7A 显示了它的代表性电流轨迹.在图7A 中,对照电流C 的大小为6 500 nA,在1 μmol·L-1的RgIA 条件下,ACh 脉冲产生的电流几乎为0 nA,其电流几乎全部被RgIA 所阻断,即图7A 中箭头所指的第一个电流轨迹.当用钡离子ND96 灌流液取代普通ND96 灌流液时,1 μmol·L-1的α -芋螺毒素RgIA 对α 9 α 10 nAChR 仍然显示出极强的阻断活性(见图7B),α 9 α 10 nAChR 的电流被完全阻断,其结果与在普通ND96 中的一样. 只是在图7B 中,对照电流C 的大小仅为250 nA,在1 μmol·L-1的RgIA 条件下,ACh脉冲产生的电流也几乎为0 nA,即图7B 中箭头所指的电流轨迹.无论是在普通ND96 灌流液中,还是在钡离子ND96 灌流液中,1 μmol·L-1的RgIA 的洗脱都是很快的,都是α 9 α 10 nAChR 的有效阻断剂,并不会受钙离子激活所产生的氯离子电流的影响.
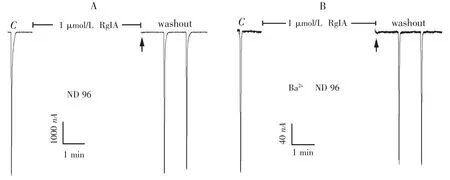
图7 1 μmol·L -1 α-芋螺毒素RgIA 在普通ND96 灌流液和钡离子ND96 灌流液中对α 9 α 10 nAChR 的电流影响
3 讨 论
本研究检测了2 个ω -芋螺毒素MVIIA 和MVIIC 对α 9 α 10nAChR 的结合活性. 虽然它们在10 μmol·L-1的高浓度条件下,在普通ND96 灌流液中显示出极其微弱或一定的阻断活性,特别是MVIIC 可阻断α 9 α 10nAChR 一半(约50%)的电流(见图3 ―4),但是,它们在不含钙离子的钡离子ND96 灌流液中,对α 9 α 10 nAChR 却完全没有阻断活性.但加入钙离子后,即将钡离子ND96 灌流液更换为普通ND96灌流液后,MVIIA 和MVIIC 又显示出微弱的阻断活性,分别与图3 和图4 的情形相似.而阳性对照α -芋螺毒素RgIA,无论是在普通ND96 灌流液中,还是在钡离子ND96 灌流液中,都能有效地阻断α 9 α 10 nAChR 的电流,它不受钙离子激活产生的氯离子电流的影响,是α 9 α 10 nAChR 的特异阻断剂(见图7).这更进一步地证实了,这2 个ω-芋螺毒素MVIIA 和MVIIC 对α 9 α 10 nAChR 的确没有抑制作用,而在常规的ND96 灌流液中它却表现出微弱的抑制活性,这是受钙离子激活产生的氯离子电流所干扰的结果.因此,这就可以回答表1 中所呈现的2 个问题了,即这2 个ω-芋螺毒素MVIIA 和MVIIC 并不是α 9 α 10 nAChR 的有效阻断剂,其作用机理与2 个α-芋螺毒素Vc1.1 和RgIA 是完全不同的.
nAChRs 是由5 个亚基组成的五聚体跨膜蛋白,分为肌肉型和神经型两大类,其中神经型nAChRs 异常复杂,它们由不同的亚基(2 ~10 个)或β(β2– β4)亚基组成异源或同源型的功能性受体亚型,这些亚型的药理学功能截然不同[24].其中,存在于外周神经系统的α 9 α 10 nAChR 备受关注,它是治疗神经痛的药物靶点,其特异阻断剂α -芋螺毒素Vc1.1 和RgIA,可以通过肌肉注射途径给药[6,16],同时,它们还能阻断由GABAB 受体介导的N-型钙离子通道,但它们与GABAB 受体和N-型钙离子通道相互作用的详细机制目前还不清楚,因而α-芋螺毒素Vc1.1 和RgIA 的镇痛机理更为复杂[20].可是反过来,阻断N-型钙通道的ω-芋螺毒素MVIIA 和MVIIC 对910 nAChR 却没有活性,它们相互之间没有联系.ω -芋螺毒素MVIIA 镇痛药物作用于仅存于中枢神经系统的N-型钙通道,需通过编程泵内置于人体内给药至脊髓,给药途径很麻烦,该给药泵非常昂贵[25]. 因此,本研究结果将有助于阐明芋螺毒素抑制α 9 α 10nAChR 和钙离子通道的机理,这对于研发新型的芋螺毒素镇痛药和更好地治疗慢性疼痛具有重要的意义.
[1]LEWIS R J,DUTERTRE S,VETTER I,et al. Conus venom peptide pharmacology[J]. Pharmacol Rev,2012,64(2):259-298.
[2]LUO S,CHRISTENSEN S,ZHANGSUN D,et al. A novel inhibitor of alpha9alpha10 nicotinic acetylcholine receptors from Conus vexillum delineates a new conotoxin superfamily[J]. PLoS One,2013,8(1):e54648.
[3]KAAS Q,YU R,JIN A H,et al. ConoServer:updated content,knowledge,and discovery tools in the conopeptide database[J]. Nucleic Acids Res,2012,40(Database issue):D325 -330.
[4]YE M,KHOO K K,XU S,et al. A helical conotoxin from Conus imperialis has a novel cysteine framework and defines a new superfamily[J]. J Biol Chem,2012,287(18):14 973 -14 983.
[5]ADAMS D J,CALLAGHAN B,BERECKI G. Analgesic conotoxins:block and G protein-coupled receptor modulation of Ntype (Ca(V)2.2)calcium channels[J]. Br J Pharmacol,2012,166(2):486 -500.
[6]MCINTOSH J M,ABSALOM N,CHEBIB M,et al. Alpha9 nicotinic acetylcholine receptors and the treatment of pain[J].Biochem Pharmacol,2009,78(7):693 -702.
[7]PHILLIPS C J. Economic burden of chronic pain[J]. Expert Rev Pharmacoecon Outcomes Res,2006,6(5):591 -601.
[8]CAMPBELL J N,MEYER R A. Mechanisms of neuropathic pain[J]. Neuron,2006,52(1):77 -92.
[9]GASKIN D J,RICHARD P. The economic costs of pain in the United States[J]. J Pain,2012,13(8):715 -724.
[10]PARK J,LUO Z D. Calcium channel functions in pain processing[J]. Channels (Austin),2010,4(6):510 -517.
[11]PEXTON T,MOELLER-BERTRAM T,SCHILLING J M,et al. Targeting voltage-gated calcium channels for the treatment of neuropathic pain:a review of drug development[J]. Expert Opin Investig Drugs,2011,20(9):1 277 -1 284.
[12]SNUTCH T P. Targeting chronic and neuropathic pain:the N-type calcium channel comes of age[J]. NeuroRx,2005,2(4):662 -670.
[13]ZAMPONI G W,LEWIS R J,TODOROVIC S M,et al. Role of voltage-gated calcium channels in ascending pain pathways[J]. Brain Res Rev,2009,60(1):84 -89.
[14]MCMAHON S B,CAFFERTY W B,MARCHAND F. Immune and glial cell factors as pain mediators and modulators[J].Exp Neurol,2005,192(2):444 -462.
[15]VINCLER M,MCINTOSH J M. Targeting the alpha9alpha10 nicotinic acetylcholine receptor to treat severe pain[J]. Expert Opin Ther Targets,2007,11(7):891 -897.
[16]VINCLER M,WITTENAUER S,PARKER R,et al. Molecular mechanism for analgesia involving specific antagonism of alpha9alpha10 nicotinic acetylcholine receptors[J]. Proc Natl Acad Sci U S A,2006,103(47):17 880 -17 884.
[17]SCHMIDTKO A,LOTSCH J,FREYNHAGEN R,et al. Ziconotide for treatment of severe chronic pain[J]. Lancet,2010,375(9725):1 569 -1 577.
[18]HILLYARD D R,MONJE V D,MINTZ I M,et al. A new Conus peptide ligand for mammalian presynaptic Ca2+channels[J]. Neuron,1992,9(1):69 -77.
[19]GOHIL K,BELL J R,RAMACHANDRAN J,et al. Neuroanatomical distribution of receptors for a novel voltage-sensitive calcium-channel antagonist,SNX-230 (omega-conopeptide MVIIC)[J]. Brain Res,1994,653(1 -2):258 -266.
[20]CUNY H,DE FAOITE A,HUYNH T G,et al. gamma-aminobutyric acid type B(GABAB)receptor expression is needed for inhibition of N-type (Cav2.2)calcium channels by analgesic alpha-conotoxins[J]. J Biol Chem,2012,287(28):23 948-23 957.
[21]FANG L,SHEN L,YU J,et al. Optimization of receptors expressed on Xenopus laevis oocytes[J].Biotech. Bull. 2013,6,167 -171.
[22]KATZ E,VERBITSKY M,ROTHLIN C V,et al. High calcium permeability and calcium block of the alpha9 nicotinic acetylcholine receptor[J].Hear Res,2000,141(1/2):117 -128.
[23]ELGOYHEN A B,VETTER D E,KATZ E,et al.alpha10:a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells[J].Proc Natl Acad Sci U S A,2001,98(6):3 501 -3 506.
[24]GOTTI C,CLEMENTI F. Neuronal nicotinic receptors:from structure to pathology[J]. Prog Neurobiol,2004,74(6):363-396.
[25]SANFORD M. Intrathecal ziconotide:a review of its use in patients with chronic pain refractory to other systemic or intrathecal analgesics[J]. CNS Drugs,2013,27(11):989 -1 002.
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
中国畜牧杂志(2021年5期)2021-12-05
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
中华养生保健(2020年7期)2020-11-16
中国畜牧杂志(2020年6期)2020-07-11
河南畜牧兽医(2017年8期)2017-11-24
上海农业学报(2017年3期)2017-04-10
西藏科技(2015年9期)2015-09-26
中国当代医药(2015年10期)2015-03-01