
大气14CO2的时空分异特征及其在化石源CO2示踪中的应用
2014-07-02 00:26牛振川周卫健吴书刚卢雪峰熊晓虎霍雯雯
地球环境学报 2014年1期
牛振川,周卫健,,吴书刚,卢雪峰,程 鹏,熊晓虎,杜 花,霍雯雯
(1.中国科学院地球环境研究所 黄土与第四纪地质国家重点实验室,西安 710075;2. 国家加速器质谱中心(西安),西安 710054;3. 西安交通大学,西安 710049)
大气14CO2的时空分异特征及其在化石源CO2示踪中的应用
牛振川1,2,周卫健1,2,3,吴书刚1,2,卢雪峰1,2,程 鹏1,2,熊晓虎1,2,杜 花1,2,霍雯雯1,2
(1.中国科学院地球环境研究所 黄土与第四纪地质国家重点实验室,西安 710075;2. 国家加速器质谱中心(西安),西安 710054;3. 西安交通大学,西安 710049)
大气CO2中放射性碳同位素(14C)的水平可以反映化石源CO2的影响程度,这对于评估我国目前化石源CO2的排放状况和制定节能减排政策具有重要的指导意义。本文在概述大气14CO2采样和分析方法的基础上,简要介绍了大气14CO2观测的起源和主要的源汇过程,重点论述了大气14CO2的时空分异特征及其驱动因素;阐述了化石源CO2浓度的估算方法及14CO2在国内外化石源CO2示踪中的应用现状,并对大气14CO2观测在我国化石源CO2示踪中的应用前景进行了展望;旨在为我国正确地开展大气14CO2的观测研究,深刻地理解特定区域大气14CO2的时空分异特征和化石源CO2的分布状况提供参考。
大气14CO2;同位素示踪;化石源CO2;时空分异特征;驱动因素
大量化石燃料的燃烧是导致自工业革命以来大气CO2浓度急剧升高的原因,也是造成全球变暖的主要因素之一(Rosa and Ribeiro,2001)。气候变暖必然引起区域水分循环的变化,导致水资源在时空上的重新分配,进而影响全球生态系统和人类生活的方方面面(蓝永超等,2008)。为了应对气候变暖,联合国气候变化框架公约参加国三次会议通过了《京都议定书》,旨在将大气中温室气体的含量稳定在一个适当的水平,进而防止剧烈的气候改变对人类造成伤害。作为CO2的排放大国和《京都议定书》的缔约国,我国面临越来越大的国际减排压力。如何科学、准确、有效地评估我国目前大气化石源CO2排放现状,不仅是一个亟待解决的环境外交问题,而且是一个重要的科学问题。相对于放射性碳同位素(14C)的半衰期5730年,化石燃料漫长的形成过程(通常几百万年以上)使其中的14C已经耗竭,因此,化石源和非化石源CO2的14C组成差异可以为评估大气化石源CO2的现状提供准确、有效的方法。本文简要介绍了大气14CO2的采样和分析方法,详细论述了大气14CO2的时空分异特征及其驱动因素,回顾了大气14CO2观测的起源及其在国外化石源CO2示踪中的应用现状,最后对大气14CO2观测在我国化石源CO2示踪中的应用前景进行了展望。
1 大气14CO2的研究方法
1.1 大气14CO2的采样方法
大气14CO2的采样方法因14C分析方法的不断改进和提高而发展和变化。早期大气14CO2分析需要较多的样品量,NaOH吸收法和分子筛吸附法是此时比较常用的两种方法。Levin et al(1980)使用小型气泵将大约15 m3的大气通过4n NaOH吸收液,利用NaOH与CO2的反应将CO2从大气中分离,最后滴加40%的硫酸溶液使Na2CO3分解将富集的CO2释放出来。Kuc(1991)、Kuc and Zimnoch(1998)使大约15 m3的大气通过含有分子筛的容器,将吸附的CO2热解后,最后得到5 dm3左右的CO2。但这两种采样方法费时、所需气体量大,且在采样过程中易污染。随着加速器质谱测量技术的逐渐使用,大气14CO2分析所需要的样品量由过去的十几m3减少到如今的几dm3。不易造成样品污染的长颈瓶(f ask)被逐渐用于大气14CO2的采样(Nakamura et al,1992;Turnbull et al,2009a;Miller et al,2012;丁平等,2013)。长颈瓶使用前预先抽真空,通过手动阀门的关闭来完成CO2气体的采集,但长颈瓶也存在易碎和不易运输的缺点,尤其是在进行大规模的采样时,耐酸碱、惰性强的Tef on气袋似乎是一种更方便的气体采样容器。研究表明,采集大气样品的复合膜气袋放置20天后,袋内CO2浓度变化小于2 ppm;且采样时应先打开气袋再开气泵,采样结束后,先关闭气泵再关气袋(顾帅,2009)。Zhou et al(2014)在预先装有磷酸溶液(pH=2)的密封f ask瓶内插入两根导管,一根为进气管,另一根为有流量控制阀的液体管,然后将f ask瓶倒立,利用流出液体产生的内外压力差自动将气体收集到f ask瓶内,气体的收集速率通过液体管上的流量控制阀来调节,具有自动、简便的优点。
由于大气CO2的采样和14C分析费时费力且花费巨大,大气14CO2的长期观测目前只在世界上有限的背景站开展,较少涉及离人类活动较近的区域。为了更经济方便地研究大气14CO2的区域分布状况,一年生植物叶片常用来代替大气CO2样品(Hsueh et al,2007;Riley et al,2008;奚娴婷等,2011)。与瞬时的大气样品相比,植物样品能完整地记录其生长期内大气14CO2的平均变化情况;且植物样品较易采集,通过对同种植物的比较更易扩展研究区域。
1.2 大气14CO2的分析方法
14C的分析技术经历了衰变计数法和加速器质谱法两个阶段。加速器质谱法是14C分析技术的一次飞跃,它综合了加速器技术、核探测技术和质谱技术,直接测定样品中的放射性同位素粒子,测量灵敏度高,最小可测量同位素比值可达10−16,14C/12C的测量精度可达0.2%。加速器质谱法所需的样品量比传统的衰变计数法要小很多,只需1 mg,可以满足C含量较低的大气样品的测量需求;且加速器质谱法的样品分析速度快,所需时间不到1小时(周卫健等,2007)。
14CO2加速器质谱的简要分析过程如下:将f ask瓶内或分子筛吸附的CO2释放到真空系统后,通过低温液氮冷阱(−196°C)和液氮+酒精冷阱(−80°C)将样品纯化,然后使用Zn–Fe法将纯化的CO2还原成石墨靶,最后在加速器质谱仪上进行14C测定。
1.3 大气14CO2的表示方法
大气CO2中14C的含量通常用Δ14C表示,其定义为:

(14C/12C)SN是指样品经过同位素质量分馏校正后的14C的放射性比值,(14C/12C)abs是指经过同位素质量分馏校正和衰变校正到1950年的绝对国际(现代碳)标准的14C的放射性比值。
2 大气14CO2观测的起源和主要的源汇
2.1 大气14CO2观测的起源
大气14CO2的观测于1954年在新西兰威灵顿地区的Makara站(41.25ºS,174.69ºE,海拔300 m)开始,1987年后气体样品的采集点搬到距离Makara站东南方向23 km的Baring Head站(41.41ºS,174.87ºE,海拔80 m)(Currie et al,2011)。随后1959年在奥地利的Vermunt站(47.06ºN,9.57ºE,海拔1800 m)、1977年在德国的Schauinsland站(47.92°N,7.92°E,海拔1205 m)、1984年在西班牙的Izñna站(28.3°N,16.48°W,海拔2400 m)、1986年在瑞士的Jungfraujoch站(46.55ºN,7.98ºE,海拔3450 m)、1987年在澳大利亚的Cape Grim站(40.68°S,144.68°E,海拔104 m)等地陆续开展了大气14CO2的观测工作(Levin et al,2010)。而1955年之前到1820年之间大气14CO2的数据常用树轮中的Δ14C来代替(Stuiver and Quay,1981)。
2.2 大气14CO2主要的源汇
大气14CO2的主要来源有核武器试验、宇宙射线和原子能工业。核武器试验共产生了100 多kmol的14C,宇宙射线14C的年产量为0.48~0.55 kmol,原子能工业14C的释放量较低,2005年约为0.09 kmol(Naegler and Levin, 2006)。大气中的14C会随着全球C循环逐渐进入海洋、陆地生物圈等C汇,而这些C汇赋存的14C在一定条件下会再回到大气中。核武器试验破坏了大气与陆地生物圈及海洋之间14C的平衡状态,陆地生物圈和海洋开始大幅度赋存大气14C,这种不平衡在1965年达到最大值550‰。核武器试验停止后,大气Δ14CO2开始下降而植物呼吸作用释放的Δ14CO2开始增长,陆地生物圈的14C汇变化趋于平缓且有逐步下降的趋势。到80年代,大气和植被之间的Δ14C又产生新的短暂平衡。90年代之后,生物呼吸作用中的Δ14CO2超过大气20‰~50‰,2005年超过15‰~40‰。陆地生物圈也因此由大气14CO2的汇转变为大气14CO2的源,其赋存的14C从1975年的约22 kmol下降到1995年19 kmol左右(Naegler and Levin,2009)。海洋14C汇赋存的14C自核武器试验后一直在增加中,是大气14CO2最主要的汇,约赋存了40.7~59.0 kmol的14C(Naegler and Levin,2006,2009)。
3 大气14CO2的时空分异特征及其驱动因素
3.1 大气14CO2的时间分异特征
3.1.1 大气14CO2的年际变化
自工业革命以来,由于化石燃料燃烧所产生的不含14C的CO2被释放到大气中,树轮记录的Δ14C显示,大气Δ14CO2开始逐步下降,到1950年大气Δ14CO2仅为−25‰左右(Stuiver and Quay,1981)。20世纪50年代中期之后由于大规模的大气核武器试验,大气Δ14CO2开始急剧增长,1955—1964年全球大气Δ14CO2平均每年增长80‰左右。北半球大气Δ14CO2的平均值在1964年达到峰值(785±62)‰(Hua and Barbetti,2004),个别观测站的Δ14CO2甚至接近1000‰(Levin et al,2003);而南半球由于大气环流的滞后,大气Δ14CO2的平均值在1965年达到峰值(635±4)‰(Hua and Barbetti,2004),其中威灵顿站在1965年达到最高值(694.5±3.9)‰(Currie et al,2011)。所以最近200年来,受人类活动的影响,大气Δ14CO2表现出显著的年际变化趋势。
1963年禁止大气核武器试验条约签订后,先前由核武器试验产生的大量14C随着全球C循环逐渐进入海洋和生物圈等C库,以及20世纪50年代起全球化石源CO2排放量的持续快速增长(Boden et al,2009),使得大气Δ14CO2又开始以近乎指数的形式下降,1964—2000年全球大气Δ14CO2平均每年下降17‰左右。到2000年,北半球Jungfraujoch和Schauinsland两站的Δ14CO2分别下降到(88.5±2.8)‰和(86.2±1.8)‰(Levin et al,2013),南半球威灵顿站降到(85.7±7.7)‰(Currie et al,2011)。之后,大气Δ14CO2的下降速度趋于平缓,到2005年威灵顿站大气Δ14CO2为(72.5±4.8)‰(Currie et al,2011);2012年Schauinsland站的Δ14CO2仅为(31.3±2.7)‰,其在2000—2012年每年约下降4.8‰(Levin et al,2013)。
3.1.2 大气14CO2的季节变化
多年的观测结果表明,大气Δ14CO2具有季节变化趋势(Levin et al,1985,1989,2010,2013;Randerson et al,2002;Currie et al,2011)。北半球大气Δ14CO2的季节变化主要受化石燃烧、生物源排放和平流层–对流层交换(Stratospheric-Tropospheric Exchange,STE)的影响,南半球的季节变化主要受海洋、化石燃烧和平流层–对流层交换的影响(Randerson et al,2002)。
北半球的观测数据表明,大气Δ14CO2高值出现在夏末秋初,70年代之前主要受平流层大气核武试验产生的14C与对流层的交换所影响,80年代之后生物源排放的14C也产生了重要作用;低值出现冬末春初,与化石源排放的CO2有关(Levin and Kromer,2004;Levin et al,1985,1989,2010,2013;Randerson et al,2002)。Vermunt站1959—1982年的观测数据表明,季节差异从1959年的60‰左右开始增长,到1963年季节差异超过300‰,之后季节差异开始下降,到70—80年代季节差异为20‰左右(Levin et al,1985)。80年之后北半球大气Δ14CO2的季节差异逐渐变小,Jungfraujoch站在1986—2012年大气Δ14CO2的季节差异仅为5‰左右(Levin et al,2010,2013),Alert站1995—2005年的季节差异为7‰(Levin et al,2010),Izñna站仅为3‰,但9月会有轻微的谷值出现(Levin et al,2010)。
南半球威灵顿站1954—2005年长期的观测结果显示,1954—1980年大气Δ14CO2在南半球的夏季较高,而在南半球的冬季较低,季节差异在1966年达到最大值(20‰)(Currie et al,2011);而1980—1989年大气Δ14CO2的季节变化规律发生转变,即南半球冬季的Δ14CO2值较高,而南半球夏末秋初的Δ14CO2较低,季节差异为3.5‰(Currie et al,2011),这种转变可能主要跟海洋和陆地14C库的影响有关(Manning et al,1990);而1990年之后Δ14CO2的季节差异不明显(Currie et al,2011)。南半球Cape Grim站1995—2005年Δ14CO2的季节差异也仅为2‰,另外一些站也无明显的季节差异(Levin et al,2010)。
3.1.3 大气14CO2的日变化
由于受经费和精力等各方面的限制,世界各地观测站大多以一到两周为间隔观测大气Δ14CO2,对其日变化而少有报道,高时间分辨率的大气Δ14CO2观测是未来值得关注的研究对象。Zondervan and Meijer(1996)在荷兰Kollumerwaard站1994年11月23日的观测结果表明,大气Δ14CO2的谷值出现在早上的7点和下午的6点,峰值出现在下午的3点和夜晚的11点,这种变化主要与早晚上下班高峰的车流量、日间工厂排放情况以及植物在日间的光合作用和夜间的呼吸作用有关。
3.2 大气14CO2的空间分异特征
大气Δ14CO2不仅具有时间上的变化趋势,而且具有空间上的变化趋势,如南北梯度、纬度差异、经度差异以及垂直梯度。北半球大气Δ14CO2的空间差异主要是受化石源CO2影响,南半球则受海气交换影响较大(Turnbull et al,2009b)。
3.2.1 大气14CO2的南北梯度
20世纪50年代中期到60年代中期,由于大气核武试验主要发生在北半球而海洋对大气Δ14CO2的吸收作用主要发生在南半球,使得北半球大气的Δ14CO2比南半球高300‰以上,之后这种差异大幅度缩小。80年代后期至今的观测数据表明,南半球大气的Δ14CO2反而比北半球高千分之几(Levin et al,2010),这跟南半球化石源CO2的浓度比北半球低3~4 ppm(GlobalView-CO2,2008)有关。
3.2.2 大气14CO2的纬度差异
对于南半球而言,中高纬度地区大气Δ14CO2的值较低,比赤道地区低2‰ ~5‰(Rozanski et al,1995),这跟南极周边14C贫化海水的上升流与大气的交换有关(Rozanski et al,1995;Levin et al,2010);南极地区具有最高的大气Δ14CO2值,比南北半球其他地区高3‰~8‰,这与南极地区宇宙射线产生的14C随平流层气团的输入有关(Levin et al,2010)。对于北半球而言,赤道地区大气Δ14CO2的值也较高,中纬度地区大气Δ14CO2的值要低2‰左右,与此地区大量的化石源CO2排放有关,而到高纬度地区,大气Δ14CO2又略微上升1‰左右(Levin et al,2010)。
3.2.3 大气14CO2的经度差异
与大气Δ14CO2的纬度差异相比,对其经度差异的关注相对较少。Turnbull et al(2009a)于2004年3—4月沿欧亚大陆(51.5 ~58.5°N)的观测结果表明,大气Δ14CO2具有从俄罗斯西部(40°E)到西伯利亚东部(120°E)升高的趋势,这种经度差异与核电厂排放的14C和化石源排放的CO2有关。而南半球纬度相似的威灵顿站和Cape Grim站1987—2005年的观测结果表明,两站具有相同的年际变化趋势,但两站之间未观测到明显的差异(Levin et al,2007;Currie et al,2011)。
3.2.4 大气14CO2的垂直梯度
由于大气核武试验、宇宙射线在平流层产生大量的14C以及平流层较弱的垂直混合作用,使得平流层和对流层之间产生了Δ14CO2梯度。南半球平流层–对流层Δ14CO2梯度主要受平流层14C产量和海洋吸收14C强弱的控制,北半球平流层–对流层梯度受自然源14C产量和由于化石源CO2排放使得对流层大气变成14C汇强弱的控制(Levin et al,2010)。平流层–对流层交换是大气Δ14CO2季节变化的重要推动力。研究表明,北半球14CO2的平流层通量在1963年对季节变化的影响超过250‰,到1970年降到30‰左右(Randerson et al,2002)。
受宇宙射线的影响,平流层大气Δ14CO2具有随海拔高度增加而增加的趋势,在日本东北部基于探空气球的观测资料表明,大气Δ14CO2在海拔21~30 km为267‰ ~309‰,在海拔19~20 km 为134‰,平流层大气Δ14CO2比对流层高80‰ ~200‰(Nakamura et al,1992)。
受地面化石源CO2排放的影响,对流层大气Δ14CO2也具有随着高度增加而增加的趋势。Miller et al(2012)在美国东北海岸的飞机观测表明,对流层大气Δ14CO2随高度的增加而增加,冬季海拔4 km的Δ14CO2比地面高20‰左右,夏季高15‰左右。Graven et al(2009)夏季在美国科罗拉多州的飞机观测结果表明,城区和郊区对流层大气Δ14CO2均具有随海拔高度而增加的趋势,且同一高度的Δ14CO2在上午10点和下午2点时保持不变。
3.3 厄尔尼诺和季风对大气Δ14CO2的影响
厄尔尼诺–南方涛动事件(El Niño/Southern Oscillation,ENSO)通过减少赤道太平洋地区的上升流和增强热带雨林生物源富含14C的CO2的释放使大气Δ14CO2增加。统计资料表明,对流层大气Δ14CO2的年际正异常与厄尔尼诺–南方涛动事件相关(Dutta,2002)。厄尔尼诺–南方涛动事件会加剧大气Δ14CO2的季节差异,90年代早期,厄尔尼诺–南方涛动事件使赤道地区大气Δ14CO2的季节差异达40‰(Rozanski et al,1995)。厄尔尼诺–南方涛动事件还会加剧大气Δ14CO2区域分布的差异。1985—1989年,热带地区生物源14CO2在厄尔尼诺–南方涛动事件的影响下,被泵入其他地区,使得Gape Grim站(41°S)和Neumayer站(71°S)之间大气Δ14CO2的差值被拉大到6‰,远高于平常年份的1‰ ~3‰;同时北半球其他站大气Δ14CO2与Neumayer站(71°S)也显示了较大的差值。但在厄尔尼诺–南方涛动事件比较强烈的1997—1998年,并未观察到上述现象(Levin et al,2010)。
季风对大气Δ14CO2的空间分布具有重要的影响。1959—1963年,北半球由核武器试验产生的14C的分布依赖于被哈德里环流圈(Hadley Cell)边界和赤道辐合带(Intertropical Convergence Zone,ITCZ)的季节性位置控制的大气环流(Hua and Barbetti,2007)。当北半球气团的14C显著高于南半球时,南半球赤道附近Muna岛1951—1979年大气Δ14CO2受富含14C的亚洲冬季风的影响,而此岛以南站点主要受南半球14C贫化气团的影响(Hua et al,2012)。
414CO2在化石源CO2示踪中的应用
4.1 化石源CO2浓度的估算方法
化石源CO2的排放会造成大气14C/12C的比值下降(Suess,1955)。在目前大气CO2浓度为380 ppm的状况下,每排放1 ppm的化石源CO2,会使大气Δ14CO2下降约2.8‰(Turnbull et al,2006),每年下降12‰ ~14‰(Levin et al,2010)。因此,大气Δ14CO2的观测可以用来反映化石源CO2的排放状况。与自下而上的源清单方法相比,14C这种自上而下的同位素示踪方法可以为评估大气化石源CO2的排放状况提供新的思路。
Levin et al(1989)假定大气CO2(CO2obs)简单地由背景大气CO2(CO2bg)和化石源CO2(CO2ff)两部分组成,通过测定Δ14CO2对欧洲Heidelberg、Westerland、Schauinsland和Jungfraujoch四站大气CO2ff的浓度和季节变化进行了研究。Zondervan and Meijer(1996)在对荷兰Kollumerwaard站大气Δ14CO2的研究中,首次考虑了生物排放源CO2(CO2bio)的贡献,将CO2obs分为CO2bg、CO2ff和CO2bio三部分,根据14C质量平衡得到公式(2)和(3):
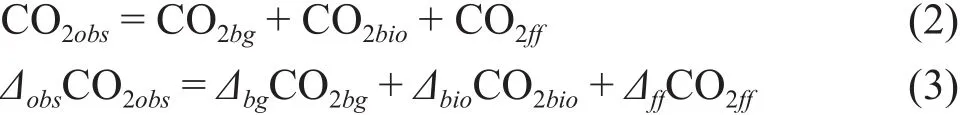
由公式(2)和(3)得到CO2ff的计算方法,
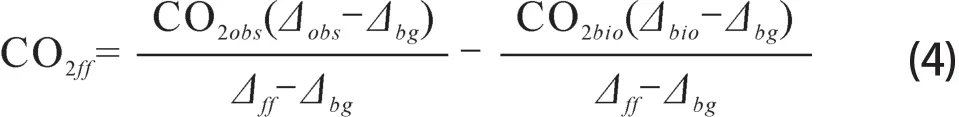
公式(4)中等号右边的第二项(β)可以写为:

Δbio可以认为近似地等于Δbg(Kuc et al,2007;Riley et al,2008;Turnbull et al,2009a;奚娴婷等,2011),则公式(4)简化为:
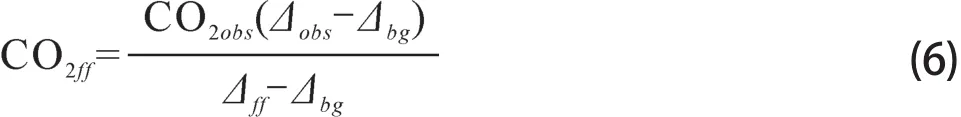
这种简化会使CO2ff的计算结果产生一定的误差,Turnbull et al(2006,2009b)在冬季CO2bio的浓度为2±1 ppm,夏季为5±2.5 ppm的情况下,估计此简化带来的误差冬季为0.2~0.3 ppm,夏季为0.4~0.8 ppm。
公式(6)中Δbg为背景大气14CO2的Δ值,理想的大气背景点是3~5 km的自由对流层,背景点太高会使14CO2受到宇宙射线产生的14C的影响(Turnbull et al,2009b),一些高山和海洋背景站也可以作为自由对流层的代表,如Jungfraujoch和威灵顿站等(Levin and Kromer,2004;Currie et al,2011)。
4.2 国外化石源CO2的研究现状
CO2浓度的三分法被普遍用在以后的化石源CO2研究中(Levin et al,2003,2008;Turnbull et al,2006,2009b;Hsueh et al,2007;Kuc et al,2007;Riley et al,2008;Graven et al,2009;Miller et al,2012),国外对化石源CO2的季节变化、日变化以及区域分布进行了大量研究。
化石源CO2具有冬季高和夏季低的特点,这与排放源的季节变化和大气扩散状况有关。Levin et al(2003,2008)的研究结果表明,1986—2006年期间Schauinsland站化石源CO2浓度为1.31±0.09 ppm,Heidelberg站为10.96±0.20 ppm;冬季Schauinsland和Heidelberg两站化石源CO2浓度分别为1.7 ppm和14 ppm,而夏季分别为1.0 ppm和6.5 ppm。
由于缺乏高分辨率的大气Δ14CO2数据,Levin and Karstens(2007)结合Δ14CO2每周的值、CO2:CO比值的周平均值以及CO的小时浓度均值,间接地对德国Heidelberg地区化石源CO2的日变化状况进行了研究,并与实地观测研究进行了验证,这种间接方法的不确定性随化石源比重的降低而升高,在欧洲大陆为15% ~ 40%。Graven et al(2009)用此方法对美国科罗拉多州化石源和生物源CO2的贡献进行了分析,早晨在郊区和城区均体现出生物呼吸作用排放的CO2的影响,午后大气CO2则主要受化石源排放和边界层高度的影响。
一年生植物叶片常被用于代替大气CO2样品,来快速、方便和经济地示踪化石源CO2的区域分布状况。Hsueh et al(2007)通过测定玉米叶中的14C对美国北部区域化石源CO2的分布状况进行了研究,其中东北部化石源CO2浓度为2.7±1.5 ppm,俄亥俄—马里兰州区域为4.3±1.0 ppm。Riley et al(2008)等通过对加利福尼亚地区128个地点C3草类中Δ14C的监测,结合大气模型模拟了此地区化石源CO2的排放通量和区域传输,洛杉矶、旧金山、中央谷地和北部海岸地区化石源CO2的浓度分别为13.7 ppm、6.1 ppm、4.8 ppm和0.3 ppm,化石源CO2由旧金山和洛杉矶向中央谷地流动,每年加州地区化石源CO2的39%、5%、35%和21%分别向东、西、南和北四个方向传输。
5 我国大气14CO2的研究现状和展望
与国外相比,我国对大气CO2和14CO2的观测研究起步较晚,自1989年在青海省瓦里关山建立我国第一个全球大气本底监测站之后(温玉璞,1997;Zhou et al,2005),相继在北京城区(王跃思,2002;王长科,2003;刘强,2005)和上甸子以及浙江临安、黑龙江龙凤山(刘立新,2009)等地开展了大气CO2的观测,但基于当时科研条件的限制,大多数研究仅限于认识大气CO2浓度随时间的变化趋势,少有涉及14CO2的观测及化石源CO2示踪的研究,目前仅在北京(奚娴婷等,2011)、广州(丁平等,2013)和西安(Zhou et al,2014)开始了初步的相关研究。
半个多世纪以来,大气14CO2在采样和分析方法、时空分异特征、主要源汇过程以及化石源CO2示踪中取得了一系列重要进展,但仍有一些复杂的科学问题有待解决;且大气14CO2研究目前主要集中在少数发达国家的有限区域内,而在广大发展中国家少有涉及,这会影响对全球化石源CO2分布状况的全面认识和国际碳减排责任的分担。结合我国的实际状况,未来我国14CO2研究可能需要在以下几方面得到加强。
(1)我国幅员辽阔、区域经济发展不平衡,而大气14CO2的观测数据又十分缺乏。因此既要注重面上的考察工作,又要建立涵盖内陆、沿海、城市、农村和背景点等不同类型地点的观测网络,以期全面认识我国大气14CO2的分布状况。
(2)在人类活动强烈的城市区域,需要高时间分辨率的大气14CO2数据来研究城市化石源CO2的变化趋势。
(3)与国外相比,我国缺乏大气14CO2的长期定位研究,建议早日在背景地区开展大气14CO2的长期观测研究,以期在国际大气14CO2数据网中占有一席之地。
(4)建议观测数据与模式模拟相结合,开展化石源CO2区域分布与传输的研究。
(5)在现有的研究方法中,笼统地将大气CO2分为CO2bg、CO2ff和CO2bio三部分,如何进一步细分化石源,即燃煤和机动车尾气的分担各自是多少,对于将来制定具体的减排政策更具有指导意义。
(6)生物呼吸作用包括自养呼吸和异养呼吸,而异养呼吸作用碳源的新老组成可能是比较复杂的。在现有的研究方法中,生物源CO2的Δ14C值是否等于背景点CO2的Δ14C值仍是一个值得商榷的科学问题。
(7)尽管目前全球大气CO2浓度的增长是由化石源CO2的排放造成的,但最新的研究结果显示,大气CO2浓度的季节和日变化更依赖于陆地生物圈(Miller et al,2012)。因此,生物源CO2也是未来一个值得研究的重要对象。
丁 平, 沈承德, 易惟熙, 等. 2013. 大气14CO2观测:2010—2011年广州城市大气中化石源CO2浓度变化特征[J]. 地球化学, 42(2): 297–306. [Ding P, Shen C D, Yi W X, et al. 2013. Fossil fuel-derived CO2contribution to the urban atmosphere in Guangzhou, South China by14CO2observation, 2010—2011 [J]. Geochimica, 42(2): 297–306.]
顾 帅. 2009.我国温室气体本底采样分析过程中的质控方法研究[D]. 北京:中国气象科学研究院,44 – 46. [Gu S. 2009. Research of quality control method of sampling and analyzing greenhouse [D]. Beijing: Chinese Academy of Meteorological Sciences, 44 – 46.]
蓝永超, 胡兴林, 肖洪浪, 等. 2008. 全球变暖情景下黑河山区水循环要素变化研究[J]. 地球科学进展, 23(7): 739–747. [Lan Y C, Hu X L, Xiao H L, et al. 2008. A study of variations of water cycle factors in the mountainous area of the Heihe’ main river under global warming scene [J]. Advances in Earth Science, 23(7): 739–747.]
刘立新, 周凌晞, 张晓春, 等. 2009. 我国4个国家级本底站大气CO2浓度变化特征 [J]. 中国科学D辑: 地球科学, 39: 222–228.[Liu L X, Zhou L X, Zhang X C, et al. 2009. The characteristics of atmospheric CO2concentration variation of four national background stations in China [J]. Science in China Series D: Earth Sciences, 39: 222–228.]
刘 强, 王跃思, 王明星, 等. 2005. 北京大气中主要温室气体近10年变化趋势 [J].大气科学, 29: 267–271.[Liu Q, Wang Y S, Wang M X, et al. 2005. Trends of greenhouse gases in recent 10 years in Beijing [J]. Chinese Journal of Atmospheric Sciences, 29: 267–271.]
王长科, 王跃思, 刘广仁. 2003. 北京城市大气CO2浓度变化特征及影响因素 [J]. 环境科学, 24: 13–17. [Wang C K, Wang Y S, Liu G R. 2003. Characteristics of atmospheric CO2variations and some affecting factors in urban area of Beijing [J]. Environmental Science, 24: 13–17.]
王跃思, 王长科, 郭雪清, 等. 2002. 北京大气CO2浓度日变化、季变化及长期趋势 [J]. 科学通报, 47: 1108–1112. [Wang Y S, Wang C K, Guo X Q, et al. 2002. Trend, seasonal and diurnal variations of atmospheric CO2in Beijing [J]. Chinese Science Bulletin, 47: 1108–1112.]
温玉璞, 汤 洁, 邵志清, 等. 1997. 瓦里关山大气二氧化碳浓度变化及地表排放影响的研究[J].应用气象学报, 8: 129–136. [Wen Y P, Tang J, Shao Z Q, et al. 1997. A study of atmospheric CO2concentration variations and emission from the soil surface at Mt. Waliguan [J]. Quarterly Journal of Applied Meteorology, 8: 129–136.]
奚娴婷, 丁杏芳, 付东坡, 等. 2011. 用一年生植物研究大气Δ14C分布与化石源CO2排放[J]. 科学通报, 56(13): 1026–1031. [Xi X T, Ding X F, Fu D P, et al. 2011. Regional14C patterns and fossil fuel derived CO2distribution study in the Beijing area using annual plants [J]. Chinese Science Bulletin, 56(13): 1026–1031.]
周卫健, 卢雪峰, 武振坤, 等. 2007. 西安加速器质谱中心多核素分析的加速器质谱仪[J].核技术, 30(8): 702–708. [Zhou W J, Lu X F, Wu Z K et al. 2007. The AMS facility at Xi'an AMS Centre [J]. Nuclear Techniques, 30(8): 702–708.]
Boden T A, Marland G, Andres R J. 2009. Global, regional, and national fossil-fuel CO2emissions [R]. Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, U.S. Department of Energy, Oak Ridge, Tenn., U.S.A. doi:10.3334/CDIAC/00001_V2010.
Currie K I, Brailsford G, Nichol S, et al. 2011. Tropospheric14CO2at Wellington, New Zealand: the world’s longest record [J]. Biogeochemistry, 104: 5–22.
Dutta K. 2002. Coherence of tropospheric14CO2with El Niño/ Southern Oscillation [J]. Geophysical Research Letters, 29(20): 1987, doi:10.1029/2002GL014753.
GlobalView-CO2. 2008. Cooperative Atmospheric Data Integration Project — Carbon Dioxide [OL] Tech. rep., NOAA ESRL, Boulder, CO, USA, available via anonymous FTP: ftp.cmdl.noaa.gov, path: ccg/CO2/ GLOBALVIEW.
Graven H D, Stephens B B, Guilderson T P, et al. 2009. Vertical profiles of biospheric and fossil fuel-derived CO2and fossil fuel CO2: CO ratios from airborne measurements of Δ14C, CO2and CO above Colorado, USA [J]. Tellus B, 61: 536–546.
Hsueh D Y, Krakauer N Y, Randerson J T, et al. 2007. Regional patterns of radiocarbon and fossil fuel-derived CO2in surface air across North America [J]. Geophysical Research Letters, 34: L02816, doi:10.1029/2006GL027032.
Hua Q, Barbetti M. 2004. Review of tropospheric bomb14C data for carbon cycle modelling and age calibration purposes [J]. Radiocarbon, 46: 1–26.
Hua Q, Barbetti M. 2007. Inf uence of atmospheric circulation on regional14CO2differences [J]. Journal of Geophysical Research, 112: D19102, doi:10.1029/2006JD007898.
Hua Q, Barbetti M, Levchenko V A, et al. 2012. Monsoonal inf uence on Southern Hemisphere14CO2[J]. Geophysical Research Letters, 39: L19806, doi:10.1029/2012GL052971.
Kuc T. 1991. Concentration and carbon isotope composition of atmospheric CO2in southern Poland [J]. Tellus B, 43: 373–378.
Kuc T, Rozanski K, Zimnoch M, et al. 2007. Two decades of regular observations of14CO2and13CO2content in atmospheric carbon dioxide in central Europe: Long-term changes of regional anthropogenic fossil CO2emissions [J]. Radiocarbon, 49: 807–816.
Kuc T, Zimnoch M. 1998. Changes of the CO2sources and sinks in a polluted urban area (southern Poland) over last decade, derived from the carbon isotope composition [J]. Radiocarbon, 40(1): 417–23.
Levin I, Hammer S, Kromer B, et al. 2008. Radiocarbon observations in atmospheric CO2: Determining fossil fuel CO2over Europe using Jungfraujoch observations as background [J]. Science of the Total Environment, 391: 211–216.
Levin I, Karstens U T E. 2007. Inferring high-resolution fossil fuel CO2records at continental sites from combined14CO2and CO observations [J]. Tellus B, 59: 245–250.
Levin I, Kromer B. 2004. The tropospheric14CO2level in midlatitudes of the Northern Hemisphere (1959—2003) [J]. Radiocarbon, 46: 1261–1272.
Levin I, Kromer B, Hammer S. 2013. Atmospheric Δ14CO2trend in Western European background air from 2000 to 2012 [J]. Tellus B, 65: 20092, http://dx.doi.org/10.3402/ tellusb.v65i0.20092.
Levin I, Kromer B, Schmidt M, et al. 2003. A novel approach for independent budgeting of fossil fuel CO2over Europe by14CO2observations [J]. Geophysical Research Letters, 30(23): 2194, doi:10.1029/2003GL018477.
Levin I, Kromer B, Schoch-Fischer H, et al. 1985. 25 years of tropospheric14C observations in central Europe [J]. Radiocarbon, 27(1): 1–19.
Levin I, Kromer B, Steele L P, et al. 2007. Continuous measurements of14C in atmospheric CO2at Cape Grim, 1997—2006 [M]// Cainey J M, Derek N, Krummel P B. Baseline Atmospheric Program Australia 2005—2006. Australian Bureau of Meteorology and CSIRO Marine and Atmospheric Research, Melbourne: 57–59.
Levin I, Miinnich K, Weiss W. 1980. The effect of anthropogenic CO2and14C sources on the distribution of14C in the atmosphere [J]. Radiocarbon, 22: 379–381.
Levin I, Naegler T, Kromer B, et al. 2010. Observations and modelling of the global distribution and long-term trend ofatmospheric14CO2[J]. Tellus B, 62: 26–46, doi:10.1111/ j.1600-0889.2009.00446.x.
Levin I, Schuchard J, Kromer B, et al. 1989. The continental European Suess effect [J]. Radiocarbon, 31: 431–440.
Manning M R, Lowe D C, Melhuish W H, et al. 1990. The use of radiocarbon measurements in atmospheric studies [J]. Radiocarbon, 32: 37–58.
Miller J B, Lehman S J, Montzka S A, et al. 2012. Linking emissions of fossil fuel CO2and other anthropogenic trace gases using atmospheric14CO2[J]. Journal of Geophysical Research, 117, D08302, doi:10.1029/2011JD017048.
Naegler T, Levin I. 2006. Closing the global radiocarbon budget 1945—2005 [J]. Journal of Geophysical Research, 111: D12311, doi:10.1029/2005JD006758.
Naegler T, Levin I. 2009. Biosphere-atmosphere gross carbon exchange flux and the Δ13CO2and Δ14CO2disequilibria constrained by the biospheric excess radiocarbon inventory [J]. Journal of Geophysical Research, 114, D17303, doi:10.1029/2008JD011116.
Nakamura T, Nakazawa T, Nakai N, et al. 1992. Measurement of14C concentrations of stratospheric CO2by accelerator mass spectrometry [J]. Radiocarbon, 34(3): 745–752.
Randerson J T, Enting I G, Schuur E A G, et al. 2002. Seasonal and latitudinal variability of troposphere Δ14CO2: Post bomb contributions from fossil fuels, oceans, the stratosphere, and the terrestrial biosphere [J]. Global Biogeochemical Cycles, 16(4): 1112, doi:10.1029/2002GB001876.
Riley W J, Hsueh D Y, Randerson J T, et al. 2008. Where do fossil fuel carbon dioxide emissions from California go? An analysis based on radiocarbon observations and an atmospheric transport model [J]. Journal of Geophysical Research, 113: G04002, doi:10.1029/2007JG000625.
Rosa L P, Ribeiro S K. 2001. The present, past, and future contributions to global warming of CO2emissions from fuels [J]. Climatic Change, 48: 289–308.
Rozanski K, Levin I, Stock J, et al. 1995. Atmospheric14CO2variations in the equatorial region [J]. Radiocarbon, 37: 509–515.
Stuiver M, Quay P. 1981. Atmospheric14C changes resulting from fossil fuel CO2release and cosmic ray flux variability [J]. Earth and Planetary Science Letters, 53: 349–362.
Suess H E. 1955. Radiocarbon concentration in modern wood [J]. Science, 122: 415.
Turnbull J C, Miller J B, Lehman S J, et al. 2006. Comparison of14CO2, CO, and SF6as tracers for recently added fossil fuel CO2in the atmosphere and implications for biological CO2exchange [J]. Geophysical Research Letters, 33: L01817, doi:10.1029/2005GL024213.
Turnbull J C, Miller J B, Lehman S J, et al. 2009a. Spatial distribution of Δ14CO2across Eurasia: measurements from the TROICA-8 expedition [J]. Atmospheric Chemistry and Physics, 9: 175–187.
Turnbull J, Rayner P, Miller J, et al. 2009b. On the use of14CO2as a tracer for fossil fuel CO2: Quantifying uncertainties using an atmospheric transport model [J]. Journal of Geophysical Research, 114, D22302, doi:10.1029/2009JD012308.
Zhou L X, Thomas J C, James W C, et al. 2005. Long-term record of atmospheric CO2and stable isotopic ratios at Waliguan Observatory: Background features and possible drivers, 1991—2002 [J]. Global Biogeochemical Cycles, 19, GB3021, doi: 10.1029/2004GB002430.
Zhou W, Wu S, Huo W, et al. 2014. Tracing fossil fuel CO2using Δ14C in Xi'an City, China [J]. Atmospheric Environment, 94: 538–545.
Zondervan A, Meijer H A J. 1996. Isotopic characterisation of CO2sources during regional pollution events using isotopic and radiocarbon analysis [J]. Tellus B, 48: 601– 612.
Spatial-temporal characteristics of atmospheric14CO2and its applications on the tracing of fossil fuel CO2
NIU Zhen-chuan1,2, ZHOU Wei-jian1,2,3, WU Shu-gang1,2, LU Xue-feng1,2, CHENG Peng1,2, XIONG Xiao-hu1,2, DU Hua1,2, HUO Wen-wen1,2
(1. State Key Laboratory of Loess and Quaternary Geology, Institute of Earth Environment, Chinese Academy of Sciences, Xi'an 710075, China; 2. National Center for Accelerator Mass Spectrometry in Xi'an, Xi'an 710054, China; 3. Xi'an Jiaotong University, Xi'an 710049, China)
The level of radiocarbon (14C) in atmospheric CO2can be used to trace the concentrations of fossil fuel CO2, which is a valuable tool for the governments to understand the current emissions of fossil fuel CO2, and then to formulate mitigation strategies for CO2emissions. In this paper, the observation history, source and sink, and collection and analysis methods of14CO2is brief y reviewed, with the emphasis on the characteristics of spatial-temporal14CO2variations and its driving forces. In addition, the method to calculate fossil fuel CO2concentration and its application on the tracing of fossil fuel CO2at abroad and home is presented, along with the suggestions for the future14CO2study in China. This paper will help to understand the characteristics of14CO2and current distributions of fossil fuel CO2in China.
atmospheric CO2; isotope tracing; fossil fuel CO2; spatial-temporal variations; driving forces
X831
A
1674-9901(2014)01-0001-09
10.7515/JEE201401001
2014-01-06
国家自然科学基金项目(41303072);中国科学院地球环境研究所黄土与第四纪地质国家重点实验室自主部署重点课题(LQ1301);中国科学院地球环境研究所青年人才项目(Y354011480)
周卫健,E-mail: weijian@loess.llqg.ac.cn
猜你喜欢
保健医苑(2022年1期)2022-08-30
家庭影院技术(2021年8期)2021-11-02
学生天地(2020年18期)2020-08-25
学生天地(2020年7期)2020-08-25
小哥白尼(神奇星球)(2020年4期)2020-07-27
河北书画研究(2016年2期)2016-08-24
小学生·多元智能大王(2015年2期)2015-05-25
小学科学(2015年2期)2015-03-11
小学科学(2015年1期)2015-03-11
科学大众·小诺贝尔(2009年9期)2009-10-23
