
X型与线型嵌段聚醚在空气/水和正庚烷/水界面上聚集行为的比较研究
2014-06-23 06:52陈贻建翟雪如徐桂英
物理化学学报 2014年1期
陈贻建 刘 腾 翟雪如 徐桂英
(山东大学胶体与界面化学教育部重点实验室,济南250100)
1 引言
PEO-PPO嵌段聚醚是一类非离子型高分子表面活性剂,其分子有着丰富的可设计性,如分子量、EO和PO含量及其比例、嵌段顺序和支化程度等.因此,该类表面活性剂的研究一直是人们感兴趣的课题.1-6支状嵌段聚醚往往具有pH敏感性、7较强的水溶性、8界面吸附能力弱9等特点.与线型聚醚相比,支状聚醚在原油破乳、10,11纳米材料合成12以及碳纳米管分散13-15等领域具有许多独特之处.而且,支状聚醚还具有毒性小、可生物降解等优点,在日化、生物医药及细胞工程等领域具有良好应用前景.16,17因此,人们对支状聚醚的研究兴趣愈来愈高.Dong等18发现,支状的Tetronic聚醚存在双pKa值现象,pKa值不依赖于分子的EO/PO比例,且它们在空气/水表面以及正己烷/水界面的吸附行为与直链的Pluronic聚醚类似.Gonzalez-Lopez等19则发现,PO/EO比例高、分子量大的Tetronic聚醚形成的胶束尺寸更大.而Mansur等8发现,EO/PO比例相近时Tetronic的水溶性好于直链的Pluronic聚醚,其原因是位于Tetronic聚醚分子中心的胺基增大了聚醚分子的亲水性,并且其支化结构使亲水的EO链分散度增大.
我们比较过七支状嵌段聚醚(AE73)20与具有相似EO/PO比例的线型嵌段聚醚Pluronic P10521的表面活性,发现EO/PO比例相近时,支状与线型聚醚分子量不同也影响其聚集行为.Washington等9也发现,支状聚醚在界面上的单分子占据面积高于线型聚醚,且由于分子结构引起的空间位阻导致其在界面上的吸附能力较弱,因此降低表面张力的效率不如线型聚醚者;但其支状结构使PO基团(尤其CH3基团)在界面上的覆盖率增大,使水表面的极性降低,因此表面张力会降低.我们还发现相同条件下AE73的扩张模量小于线型的Pluronic P123者,22例如,扩张频率为0.1 Hz时,P123在空气/水表面的扩张模量为18.3 mN·m-1,而AE73的扩张模量仅为9.4 mN·m-1.可能的原因是,线型聚醚P123分子间相互作用强于AE73者,并且位于AE73分子中心的胺基基团增大了其亲水性,降低了其在界面上的吸附能力.另外,线型聚醚比支状聚醚更容易在吸附层形成“环”“尾”结构.因此,当界面受到扰动时线型聚醚比支状聚醚会发生更加复杂的弛豫过程.这些研究结果表明,聚醚的聚集行为除了依赖于其分子量、EO和PO含量及其比例、嵌段顺序等因素外,还与其大分子链的支化程度有关.但支化程度对聚醚物理化学性质的影响规律尚不够明确.
为了系统比较支状与线型聚醚界面聚集行为的差异,我们设计合成了两种EO含量和分子量均相同的嵌段聚醚:线型和X型.通过静、动态界面张力以及界面扩张流变方法对比研究了它们在空气/水及正庚烷/水界面的聚集行为.以期为理解支状结构对嵌段聚醚聚集行为的影响机理及其在泡沫和乳状液稳定性控制等方面提供信息.
2 实验部分
2.1 药品
线性和X型嵌段聚醚分别简写为LPE和TPE,均为本实验室合成产品,其分子结构如图1所示.正庚烷、丙二醇、乙二胺和氢氧化钾为化学纯试剂,购自国药集团化学试剂有限公司,环氧丙烷和环氧乙烷购于滨州市华茂工贸有限公司.实验用水均经三次蒸馏.
2.2 实验方法
2.2.1 嵌段聚醚的合成
LPE和TPE均通过阴离子聚合反应合成(同文献22).合成时,在反应釜中加入起始剂丙二醇(或乙二胺)及催化剂氢氧化钾,抽真空半小时后加热到100°C,然后逐渐通入环氧丙烷.反应过程中控制压力和温度分别在0.3 MPa和135°C以下.随着反应的进行,压力会逐渐降低,当环氧丙烷加完后压力不再降低时,降低温度到90°C以下,通入N2将反应的中间产物排出.然后按照相同的方法通入环氧乙烷,控制压力和温度分别在0.3 MPa和120°C以下,待反应完毕即得到粗产物LPE或TPE.将粗产物溶解到适量纯水中,滴加乙酸中和氢氧化钾.并用0.45 μm的微膜过滤除去不溶性杂质.过滤后用二氯甲烷进行萃取,再通过旋转蒸发除去溶剂,得到的产物放入真空干燥箱中60°C下真空干燥24 h.
2.2.2 嵌段聚醚的EO/PO测定
LPE和TPE的EO和PO含量通过1H核磁共振(1H-NMR)测定.由于核磁共振谱图中质子峰的面积与质子的数目成正比.因此,以氘代氯仿为溶剂,用Bruker AV-400核磁共振仪测定样品的1H-NMR谱图(Supporting Information,图S1),通过3.65和1.14处质子峰的面积计算聚醚中EO和PO基团的数目,进而获得EO和PO的质量分数.设计和测量结果如表1所示.
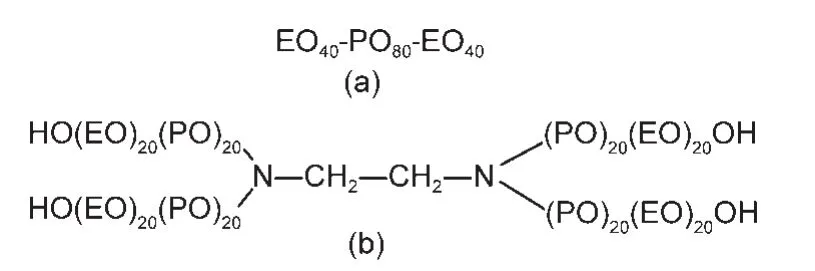
图1 LPE(a)和TPE(b)的化学结构示意图Fig.1 Scheme of chemical structures for LPE(a)and TPE(b)

表1 嵌段聚醚LPE和TPE的分子组成Table 1 Molecular composition of the block polyethers LPE and TPE
2.2.3 平衡界面张力测定
用吊片法在PROCESSOR TENSIOMETER K-12程序表面张力仪(德国Krüss公司,精度为0.01 mN·m-1)上测量一系列不同浓度聚醚溶液的表(界)面张力,试样温度用恒温槽(德国HAAKE公司,精度为0.1°C)控制,平衡时间为0.5 h,每个试样测量3次,取其平均值.
2.2.4 动态界面张力和界面扩张粘弹性
在Tracker界面流变仪(法国,Teclis公司)上利用滴外形法测定空气/水和正庚烷/水界面的动态界面张力和扩张粘弹性.通过与毛细管相连的马达控制活塞运动,使气泡(或液滴)的大小发生变化,改变界面的面积大小,并用CCD照相机采集气泡和油滴形状的变化信息,由软件计算得到界面张力值,同时通过马达做周期性运动来得到界面扩张粘弹性数据.本软件可以实时地进行计算,因此可以得到表面张力随时间的变化.测量时将不同浓度的聚醚溶液盛在石英皿中,弯头注射器中装满空气或正庚烷,然后将注射器针头插入液面以下,通过马达运动来控制注射器形成气泡或油滴从而得到空气/水表面和正庚烷/水界面,通过软件控制气泡体积的变化为总体积的10%,振荡频率为0.005-0.05 Hz.
界面张力和扩张粘弹性的测定和试样放置温度均恒定在(25.0±0.1)°C.
3 结果与讨论
3.1 不同结构嵌段聚醚的界面活性
界面活性是表面活性剂最基本也是最重要的特性之一.润湿、起泡、乳化及增溶等许多作用均与其有关.界面活性可以用界面张力(γ)等温线的斜率表示之.23-25c为界面浓度,α为界面张力等温线接近临界胶束浓度(cmc)部分的斜率.α值愈大,表示两亲分子的界面活性愈高.图2A示出了LPE和TPE水溶液的表面张力等温线.显然,它们均出现两个转折点,而且浓度较低时两者的表面张力几乎相同,但随着浓度增大,两者之间出现明显的差异.取等温线两拐点之间部分的斜率求得其α值,当LPE浓度低于4 mg·L-1时,表面张力线性下降(相关系数为0.991,α=21.21),继续增加其浓度,表面张力下降缓慢(相关系数为0.990,α=4.20),浓度达到2000 mg·L-1后再增加浓度,表面张力几乎不变;而TPE的浓度低于6 mg·L-1时,表面张力线性降低(相关系数为0.997,α=19.59),随着TPE浓度增加,表面张力继续下降(相关系数为0.981,α=3.01),当TPE浓度达5000 mg·L-1时表面张力才开始趋于恒定.这说明当分子量相近时,支状聚醚的表面活性比直链者小.这与Washington等9的结果相一致.
与小分子表面活性剂不同,高分子表面活性剂的表面张力等温线常常观察到双拐点现象.5,22通常认为,26-29表面张力等温线上第一个拐点的出现是嵌段聚醚较宽的分子量分布、分子在界面上构象的变化以及单分子胶束或寡聚体的形成所致;而其第二拐点对应的浓度才是其cmc值.由于LPE和TPE的平均分子量均接近8000 g·mol-1,且多分散系数为1.2,分子量较高者在浓度较低时就可能形成寡聚体,从而影响其界面吸附.因此两者均出现双拐点现象.若以第二拐点对应的浓度为其cmc,则LPE的cmc和γcmc分别为1300 mg·L-1和33.62 mN·m-1;而TPE的cmc和γcmc则为 5000 mg·L-1和34.49 mN·m-1.这表明LPE降低水表面张力的效率和效能均高于TPE者.
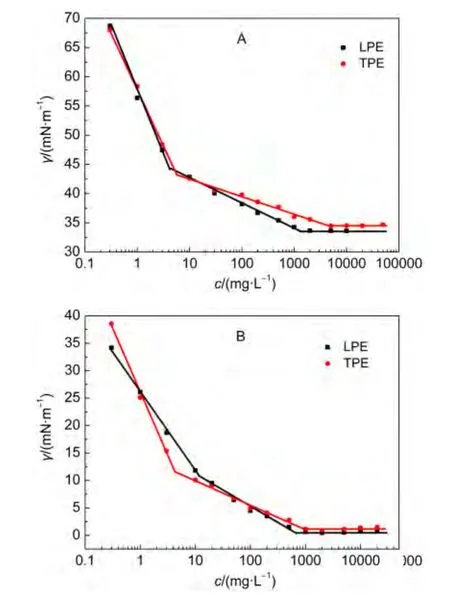
图2 298 K时LPE和TPE在空气/水表面(A)和正庚烷/水(B)界面张力(γ)等温线Fig.2 Interfacial tension(γ)isotherms of LPE and TPE at air/water(A)and n-heptane/water(B)interfaces

图3 (a,a′)线型LPE和(b,b′)支状TPE分子在空气/水表面上的吸附模型Fig.3 Schematic illustrations of possible adsorption model of(a,a′)LPE and(b,b′)TPE at air/water surface
根据Gibbs吸附等温式,可通过表面张力等温线得到分子在界面的饱和吸附量(Γmax)和最小分子占据面积(Amin).LPE和TPE在空气/水界面上的Γmax分别为6.04和4.32 mg·m-2,Amin分别为2.26和3.15 nm2.即TPE的Γmax低于LPE者,而Amin高于LPE者.LPE和TPE的分子量相同,因此可以推断,LPE分子采取更为紧密的方式聚集在液面上,所以其Γmax值较大,Amin较小;而TPE分子在液面上的聚集相对疏松,导致Amin较大,Γmax则较小.可以推测,LPE和TPE在空气/水表面上的吸附模型如图3所示:TPE分子的PO基团呈X状铺展在空气/水表面,EO链段伸向水中;而LPE分子在气/液表面呈倒U状,则PO链呈线型铺展在空气/水表面上,EO链伸向水中.30即,支状聚醚在界面上的单分子占据面积较大,且由于其分子结构引起的空间位阻导致其在界面上的吸附能力较弱,且分子间的相互作用也较小,不易在界面上紧密排列,因而表面活性低于线型聚醚者.
LPE和TPE体系的正庚烷/水界面张力等温线如图2B所示.298 K时正庚烷/水的界面张力为45.15 mN·m-1,加入LPE和TPE后可使其界面张力显著降低.显然,界面张力的变化趋势与其表面张力曲线相似,也出现双拐点现象.按照聚醚分子在表面吸附的方式处理,得到LPE和TPE的α值分别为5.56和4.48,γcmc值分别为0.63和1.10 mN·m-1,cmc值分别为650和1000 mg·L-1.显然,在正庚烷/水界面LPE的界面活性也高于TPE者.而且,比较空气/水与正庚烷/水界面的活性参数可以看出,由正庚烷/水界面得出的cmc小于由表面张力所得之值.这与正庚烷分子插入到界面层有关.31
3.2 动态界面张力
动态界面张力是研究两亲分子吸附动力学的一种常用方法,32它比平衡界面张力更能准确地反映两亲分子的扩散和吸附性能.LPE和TPE在空气/水与正庚烷/水界面的动态界面张力曲线如图4和图S2所示.由此可以看出,随着聚醚浓度增大,其在空气/水与正庚烷/水界面的界面张力γ(t)都不断减小(所测聚醚浓度均低于其cmc).动态界面张力曲线大致分为三个区域:33,34(1)诱导区,t<τ1部分;(2)界面张力快速下降区,τ1
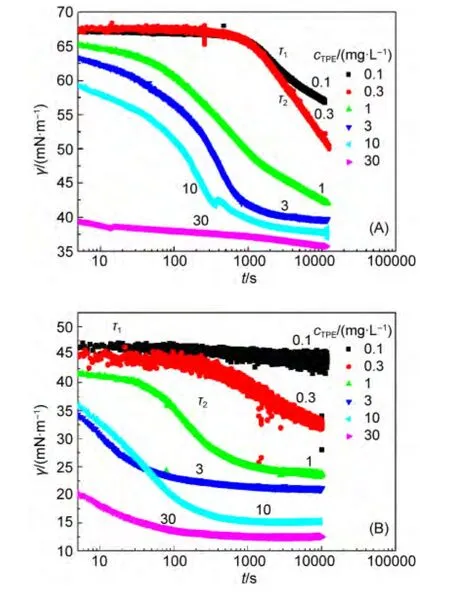
图4 不同浓度TPE的动态界面张力曲线Fig.4 Dynamic interface tension curves of TPE at various concentrations
诱导区的主要特征是,当体相浓度较低时界面张力随时间变化较缓慢或基本无变化,在此区内聚醚分子在界面上采取相对平躺的构象,较少形成“环”和“尾”结构.此时的二维吸附层结构相当疏松且具有可通透性,使得新到达的分子总能够找到“空”的吸附位点.35此区的吸附动力学主要由体相到次界面层之间的分子扩散控制.从图4和图S2可以看出,LPE和TPE在正庚烷/水界面的诱导时间小于空气/水表面者.例如,当cTPE=0.3 mg·L-1时,空气/水表面的τ1值约为2000 s,而正庚烷/水界面的τ1值约为500 s.这可能是因为PO基团与正庚烷分子的相互作用比其与空气分子的强,PO链段更容易在正庚烷/水界面吸附.界面张力快速下降区的出现可能归因于吸附分子界面浓度的显著增大.一般认为这种下降趋势持续时间较长是复杂的分子弛豫过程所致:此区内的吸附过程除了受两亲分子从体相到界面的扩散作用控制外,还与两亲分子在界面吸附层中的结构重排等弛豫过程密切相关.而TPE的支状结构和较大的分子量也是导致较长弛豫时间的原因.吸附曲线最后的介平衡或平衡区主要与已经形成的吸附层产生的能垒和空间位阻有关,此时的吸附分子在界面层呈紧密排列状态,发生较缓慢的结构重排.其中诱导区和界面张力快速下降区由扩散控制,即两亲分子的吸附过程主要是由其从体相到界面的扩散作用决定.而介平衡区界面张力下降速率较小归因于已经形成的吸附层对后来扩散到界面的分子的阻碍作用.
动态界面张力曲线可用以下方程描述:34,35

式中γ0是溶剂的表面张力,γt和γm分别是时间为t时和达到介平衡时的界面张力,n和t*是常数.n值反映了吸附初期分子从体相到次界面层的扩散过程,n值越小,扩散势垒越小,分子扩散越快,越容易达到介平衡区;t*反映了吸附后期吸附分子从次界面层到界面的吸附过程,t*值越小,吸附势垒越大,两亲分子越不易吸附到界面.两者的综合作用决定了两亲分子的吸附和扩散过程.
方程(1)线性形式可表示为:
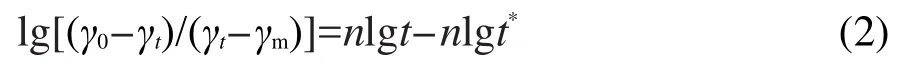
TPE之n值低于LPE者,说明其扩散较快,因为虽然LPE和TPE含有相同的PO数,但前者是线型的,分子间更容易发生相互交叠,而TPE分子有四个分支,分子间存在大的空间位阻,不易发生分子间的交叠,受其它分子的束缚力小,所以扩散反而较快.空气/水表面之n值高于正庚烷/水界面者,这可能是因为正庚烷与聚醚分子之间的疏水作用较强,正庚烷分子插入到界面层中,聚醚分子可以在正庚烷/水界面上采取更为直立状态,38,39这有利于加速聚醚分子从溶液中向界面扩散,所以n值较低.而在空气/水表面由于空气分子与聚醚分子间相互作用较弱,所以聚醚分子的构象比较无序.图5结果还表明,t*值随聚醚浓度增大而减小.在吸附后期,聚醚分子在界面上接近于饱和状态,随着浓度增加,聚醚分子的空间排斥作用导致其分子从次界面到界面层的迁移变得越来越困难.在空气/水表面之t*值比在正庚烷/水界面的大.说明聚醚分子在正庚烷/水界面的吸附更容易.TPE的t*值都较LPE的低,说明TPE分子在次界面到界面层的迁移更慢,这是由于TPE分子存在四个支链,空间位阻较大,界面上的TPE分子需要更长的时间进行构象转变,弛豫时间较长,所以t*值较低.
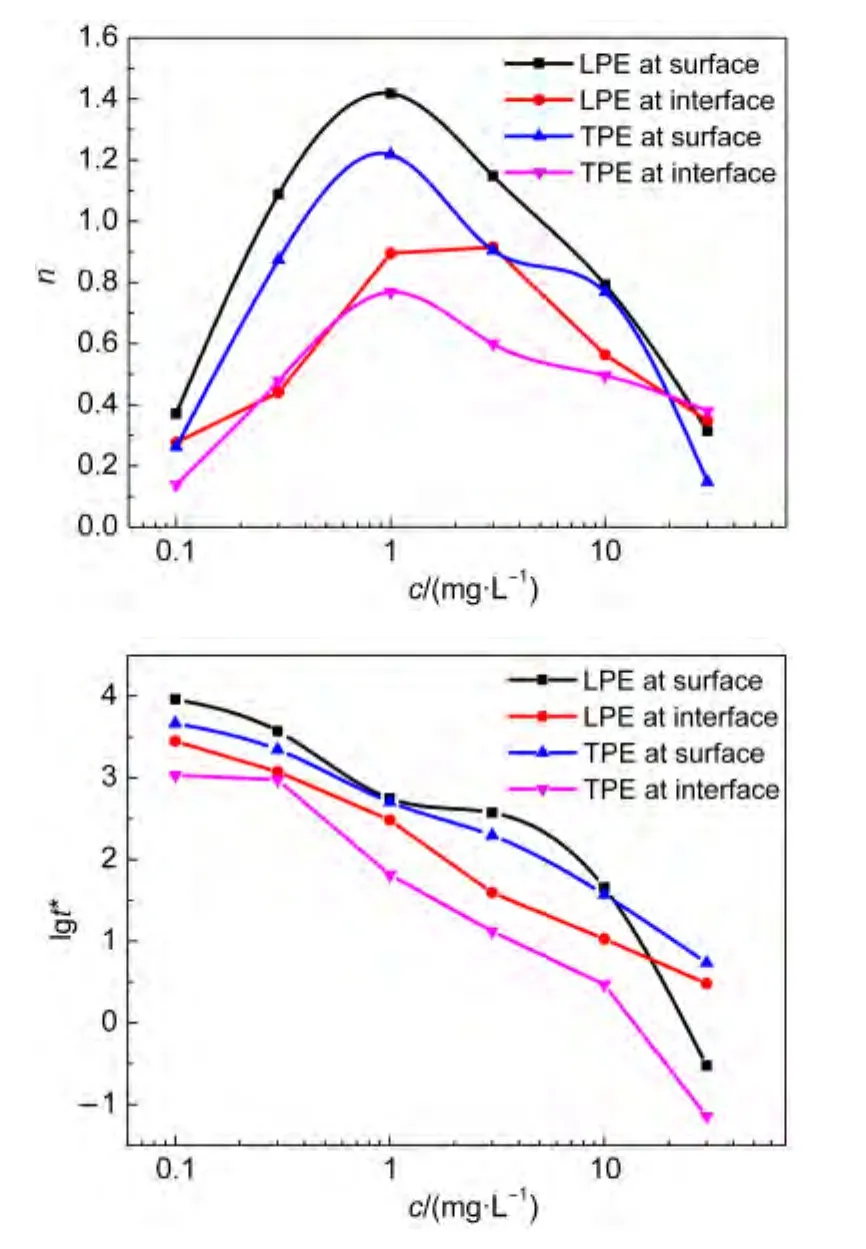
图5 Rosen方法处理得到的动态界面张力参数(t*,n)随聚醚浓度的变化Fig.5 Change of concentrations of polyethers on the parameters(t*,n)of dynamic interface tension dealt with Rosen method
3.3 界面粘弹性
通过界面扩张流变特性参数测定可以获得两亲分子在界面及其附近扩散交换、取向变化、重排、形成聚集体等微观过程的信息,对阐明泡沫或乳状液的稳定性具有重要意义.40-43图6和图S3示出了不同扩张频率下LPE和TPE在空气/水与正庚烷/水界面的扩张弹性随浓度的变化.由此可见,体系的扩张弹性随频率增加而逐渐增加.扩张弹性又称为储能模量,其来源主要是界面分子因扰动偏离平衡状态所导致的能量改变,它与分子间相互作用密切相关.频率愈高,扰动愈强,界面分子偏离平衡状态愈甚,能量变化越大,因此,扩张弹性随频率升高而增大.体系的扩张弹性随浓度增加呈现先增加后降低的趋势.当界面浓度较低时,聚醚分子不会形成环、尾结构而是以二维构象平躺在界面层.此时聚醚的扩张弹性主要由界面上相邻分子间的相互作用决定.而此种相互作用随聚醚界面浓度升高而增大,因而扩张弹性不断增大.随着聚醚浓度进一步增大,聚醚分子在界面的吸附趋于饱和,从而阻碍吸附在界面层的大分子线团的伸展.因此,聚醚分子在界面远端区形成一些环尾结构,并且引发聚醚分子在近界面区与远端区之间的弛豫,使界面扩张弹性继续增大.然而,聚醚浓度增大到一定值后,其分子在界面层不同区域、次界面层与界面层以及体相与界面层之间的弛豫交换速度加快,界面层分子恢复到“平衡态”所需的时间更短.因此,聚醚的界面扩张弹性开始降低.
为了更清楚地比较聚醚在空气/水和正庚烷/水界面扩张粘弹性的差异,绘制了频率为0.01 Hz时聚醚扩张弹性随浓度的变化(图6(C)).显然,聚醚在空气/水表面的扩张弹性随浓度的变化规律与正庚烷/水界面者相似,其扩张弹性均随聚醚浓度增加出现最大值现象.但是,在相同聚醚浓度时,与空气/水表面相比,聚醚在正庚烷/水界面的扩张模量更小并且达到最大值所需的聚醚浓度更大.可能的原因是,正庚烷与吸附在界面上的聚醚分子之间存在较强的疏水作用,因此正庚烷分子会插入界面层中聚醚分子之间,使其相邻分子之间相互作用减弱,43则吸附的聚醚分子的凝聚力减弱,造成界面弹性降低.
图6结果还表明,TPE的扩张弹性小于LPE者.扩张弹性反映界面膜恢复形变的能力,是界面膜的固态性质.TPE的扩张弹性低,表明呈X型的大分子易变形,分子间的相互作用较弱,且分子发生构象转变的弛豫时间较长,所以其恢复形变的能力弱.而LPE分子的构象转变弛豫时间较短,且分子间相互作用强,在外力作用下其恢复形变的能力也较强,因此表现出较高的扩张弹性.这与表面张力所得饱和吸附量的规律相一致.
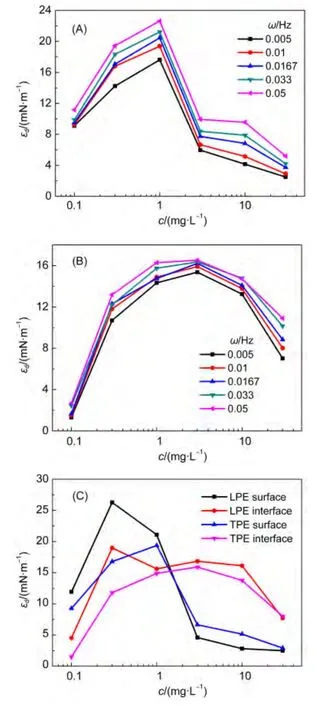
图6 不同频率(ω)下TPE体系的扩张弹性(εd)随浓度的变化Fig.6 Change of concentrations on the dilational elasticity(εd)of TPE at different frequencies(ω)

图7 不同频率(ω)下TPE体系的扩张粘性(ωηd)随浓度的变化Fig.7 Change of concentrations on the dilational viscosity(ωηd)of TPE at different frequency
研究体系的扩张粘性变化对于理解分子在界面的扩散以及构象的变化具有重要意义.44扩张粘性部分又称损耗模量,与两亲分子在界面与次界面层的交换、界面分子排布方式的改变等弛豫过程有关.45,46图7和图S4示出了不同体系的扩张粘性随浓度的变化.显然,聚醚在气/液和油/水界面的扩张粘性均显著低于其扩张弹性,说明两种聚醚在空气/水和正庚烷/水界面所形成的吸附膜均以弹性为主.而且在不同频率条件下,两种聚醚的扩张粘性均随浓度增大也出现一最大值,且出现最大值对应的聚醚浓度与扩张弹性者处于同一浓度区域.这是因为扩张粘性来源于不同的弛豫过程,而粘性与能量的耗散和重新恢复平衡状态的难易程度有关.随着浓度增大,各种弛豫过程对粘性的贡献均有不同程度增加.体相浓度增大到一定数值,弛豫过程降低扩张模量的作用更大,扩张粘性通过最大值.界面扩张粘性随扩张频率增大而减小.这是由于扩张频率越大,压缩速度越快,表面分子向体相扩散的速度越快,则表面分子向体相扩散的阻力越小,所以扩张粘性随着扩张频率的增大而减少.
以上结果还表明,TPE的扩张粘性也低于LPE者.扩张粘性反映界面膜阻滞形变的能力,是界面膜的液态性质,其大小为单位外力作用下界面膜的形变速率(即单位时间内单位外力作用下界面膜的形变),形变速率越小,扩张粘性越大.LPE形成的吸附膜中分子间相互作用强,能形成较为紧密有序的膜结构,阻滞外力作用也强,单位外力作用下界面膜的形变速率小,故扩张粘性高.而TPE分子间相互作用弱,易形变,形成膜的结构相对较为疏松,单位外力作用下界面膜的形变速率大,则其扩张粘性低.
聚醚在空气/水表面的扩张粘性大于其正庚烷/水界面者.说明正庚烷分子插入聚醚吸附层中,由于其与聚醚的PO基团有较强的相互作用使聚醚分子在界面上的排列变得相对更为有序,当正庚烷/水界面被压缩或扩张时,主要发生聚醚分子在体相和界面上的扩散交换行为;而在空气/水表面上,空气分子小且与PO基团相互作用极弱,因而聚醚分子在空气/水表面采取更加无序的分布构象.当界面层受到扰动时,除了此种扩散交换,还存在聚醚分子取向的改变,也就是说体系的弛豫行为更强,所以聚醚分子在正庚烷/水界面的扩张模量小于在空气/水表面之值.
4 结论
通过阴离子聚合方法合成了EO含量和分子量均相同的线型和X型嵌段聚醚(LPE和TPE).通过静、动态界面张力、界面扩张流变等手段对比研究了它们在空气/水以及正庚烷/水界面的聚集行为.主要得到以下结论:
(1)X型聚醚TPE降低水、正庚烷界面张力的效率和效能均低于线型聚醚LPE者.由正庚烷/水界面张力等温线所得cmc值小于由表面张力等温线所得之值.
(2)聚醚分子在正庚烷/水界面达到吸附平衡的时间比空气/水表面短,归因于正庚烷与聚醚分子之间较强的疏水作用,正庚烷分子易插入到聚醚吸附层中,因此聚醚分子在正庚烷/水界面扩散较快.而空气分子与聚醚分子间相互作用较弱,在空气/水表面上聚醚分子的构象比较无序.
(3)两种聚醚在空气/水和正庚烷/水界面形成的吸附膜均以弹性为主,X型聚醚TPE的扩张弹性比线型聚醚LPE者小,说明LPE分子之间的相互作用强,在外力作用下其恢复作用较强,而X型TPE分子间作用较弱,其恢复作用也较弱,故扩张弹性较低.
Supporting Information:available free of chargeviathe internet at http://www.whxb.pku.edu.cn.
(1) Alexandridis,P.;Hatton,T.A.Colloids Surf.A1995,96,1.doi:10.1016/0927-7757(94)03028-X
(2) Zana,R.;Marques,C.;Johner,A.Adv.Colloid Interface Sci.2006,123,345.doi:10.1016/j.cis.2006.05.011
(3) Noskov,B.A.;Lin,S.Y.;Loglio,G.;Rubio,R.G.;Miller,R.Langmuir2006,22,2647.doi:10.1021/la052662d
(4) Díez-Pascual,A.M.;Monroy,F.;Ortega,F.;Rubio,R.G.;Miller,R.;Noskov,B.A.Langmuir2007,23,3802.doi:10.1021/la062936c
(5) Gong,H.;Xu,G.;Ding,H.;Shi,X.;Tan,Y.Eur.Polym.J.2009,45,2540.doi:10.1016/j.eurpolymj.2009.05.027
(6) Shou,Q.H.;Guo,C.;Yang,L.;Jia,L.W.;Liu,C.Z.;Liu,H.Z.J.Colloid Interface Sci.2011,363,481.doi:10.1016/j.jcis.2011.07.021
(7) Kadam,Y.;Singh,K.;Marangoni,D.G.;Ma,J.H.;Aswal,V.K.;Bahadur,P.J.Colloid Interface Sci.2010,351,449.doi:10.1016/j.jcis.2010.07.046
(8) Mansur,C.R.;Barboza,S.P.;González,G.;Lucas,E.F.J.Colloid Interface Sci.2004,271,232.doi:10.1016/j.jcis.2003.11.034
(9)Washington,C.;King,S.M.;Heenan,R.K.J.Phys.Chem.1996,100,7603.doi:10.1021/jp953007p
(10)Zhang,Z.Q.;Xu,G.Y.;Wang,F.;Dong,S.L.;Chen,Y.J.J.Colloid Interface Sci.2005,282,1.doi:10.1016/j.jcis.2004.08.144
(11)Zhang,Z.Q.;Xu,G.Y.;Wang,F.;Dong,S.L.;Li,Y.M.J.Colloid Interface Sci.2004,277,464.doi:10.1016/j.jcis.2004.04.035
(12)Wang,F.;Xu,G.Y.;Zhang,Z.Q.;Xin,X.Eur.J.Inorg.Chem.2006,1,109.
(13) Xin,X.;Xu,G.Y.;Zhao,T.T.;Zhu,Y.Y.;Shi,X.F.;Gong,H.J.;Zhang,Z.Q.J.Phys.Chem.C2008,112,16377.doi:10.1021/jp8059344
(14)Gong,H.J.;Xu,G.Y.;Liu,T.;Pang,J.Y.;Dou,W.L.;Xin,X.Colloid.Polym.Sci.2011,289,933.doi:10.1007/s00396-011-2419-7
(15) Liu,T.;Xu,G.Y.;Zhang,J.;Zhang,H.H.;Pang,J.Y.Colloid.Polym.Sci.2013,291,691.doi:10.1007/s00396-012-2776-x
(16)Cao,X.R.;Xu,G.Y.;Yuan,S.L.;Gao,B.Y.Soft Matter2011,7,9035.doi:10.1039/c1sm05319a
(17) Chiappetta,D.A.;Alvarez-Lorenzo,C.;Rey-Rico,A.;Taboada,P.;Concheiro,A.;Sosnik,A.Eur.J.Pharm.Biopharm.2010,76,24.doi:10.1016/j.ejpb.2010.05.007
(18) Dong,J.F.;Chowdhry,B.Z.;Leharne,S.A.Colloids Surf.A2003,212,9.doi:10.1016/S0927-7757(02)00295-9
(19) Gonzalez-Lopez,J.;Alvarez-Lorenzo,C.;Taboada,P.;Sosnik,A.;Sandez-Macho,I.;Concheiro,A.Langmuir2008,24,10688.doi:10.1021/la8016563
(20) Zhai,X.R.;Xu,G.Y.;Chen,Y.J.;Liu,T.;Zhang,J.;Yuan,J.;Tan,Y.B.;Zhang,J.2013,291,2825.doi:10.1007/s00396-013-3013-y
(21)Alexandridis,P.;Athanassiou,V.;Hatton,T.A.Langmuir1995,11,2442.doi:10.1021/la00007a022
(22) Gong,H.J.;Xu,G.Y.;Liu,T.;Xu,L.;Zhai,X.R.;Zhang,J.;Lv,X.Langmuir2012,28,13590.doi:10.1021/la303430c
(23) Mulqueen,M.;Blankschtein,D.Langmuir2001,18,365.
(24) Kim,Y.H.;Wasan,D.T.;Breen,P.J.Colloids Surf.A1995,95,235.doi:10.1016/0927-7757(94)03032-U
(25) Aveyard,R.;Binks,B.P.;Fletcher,P.D.I.;Lu,J.R.J.Colloid Interface Sci.1990,139,128.doi:10.1016/0021-9797(90)90450-3
(26) Vieira,J.B.;Li,Z.X.;Thomas,R.K.J.Phys.Chem.B2002,106,5400.doi:10.1021/jp013286i
(27)Alexandridis,P.;Athanassiou,V.;Fukuda,S.;Hatton,T.A.Langmuir1994,10,2604.doi:10.1021/la00020a019
(28) Liu,T.;Xu,G.Y.;Gong,H.;Pang,J.;He,F.Langmuir2011,27,9253.doi:10.1021/la201676u
(29) Wanka,G.;Hoffmann,H.;Ulbricht,W.Colloid.Polym.Sci.1990,268,101.doi:10.1007/BF01513189
(30)Miller,R.;Fainerman,V.B.;Aksenenko,E.V.;Leser,M.E.;Michel,M.Langmuir2004,20,771.doi:10.1021/la030332s
(31)Zhang,H.X.;Xu,G.Y.;Wu,D.;Wang,S.W.Colloids Surf.A2008,317,289.doi:10.1016/j.colsurfa.2007.10.033
(32) Phang,T.L.;Liao,Y.C.;Franses,E.I.Langmuir2004,20,4004.doi:10.1021/la035424w
(33) Hua,X.Y.;Rosen,M.J.J.Colloid Interface Sci.1988,124,652.doi:10.1016/0021-9797(88)90203-2
(34) Rosen,M.J.;Song,L.D.J.Colloid Interface Sci.1996,179,261.doi:10.1006/jcis.1996.0212
(35) Nahringbauer,I.J.Colloid Interface Sci.1995,176,318.doi:10.1006/jcis.1995.9961
(36) Fainerman,V.B.;Lylyk,S.V.;Aksenenko,E.V.;Liggieri,L.;Makievski,A.V.;Petkov,J.T.;Yorke,J.;Miller,R.Colloids Surf.A2009,334,8.
(37)Knock,M.M.;Bell,G.R.;Hill,E.K.;Turner,H.J.;Bain,C.D.J.Phys.Chem.B2003,107,10801.doi:10.1021/jp027047m
(38) Chanda,J.;Bandyopadhyay,S.J.Phys.Chem.B2006,110,23482.doi:10.1021/jp063205o
(39) Zhang,L.;Wang,X.C.;Gong,Q.T.;Luo,L.;Zhao,S.;Yu,J.Y.J.Colloid Interface Sci.2008,327,451.doi:10.1016/j.jcis.2008.08.019
(40) Wang,Z.L.;Li,Z.Q.;Zhang,L.;Huang,H.Y.;Zhang,L.;Zhao,S.;Yu,J.Y.J.Chem.Eng.Data2011,56,2393.doi:10.1021/je1013312
(41)Zong,H.;Wang,L.;Fang,H.B.;Mao,L.T.;Wang,Y.H.;Zhang,L.;Zhao,S.;Yu,J.Y.Acta Phys.-Chim.Sin.2010,26,2982.[宗 华,王 磊,方洪波,毛雷霆,王宇慧,张 路,赵濉,俞稼镛.物理化学学报,2010,26,2982.]doi:10.3866/PKU.WHXB20101105
(42) Zhang,L.;Wang,X.C.;Gong,Q.T.;Luo,L.;Zhang,L.;Zhao,S.;Yu,J.Y.Acta Phys.-Chim.Sin.2007,23,1652.[张 磊,王晓春,宫清涛,罗 澜,张 路,赵 濉,俞稼镛.物理化学学报,2007,23,1652.]doi:10.3866/PKU.WHXB20071031
(43) Li,X.L.;Zhang,L.;Gong,Q.T.;Zhang,L.;Zhao,S.;Yu,J.Y.Acta Phys.-Chim.Sin.2010,26,631.[李秀兰,张 磊,宫清涛,张 路,赵 濉,俞稼镛.物理化学学报,2010,26,631.]doi:10.3866/PKU.WHXB20100319
(44) Noskov,B.A.Curr.Opin.Colloid Interface Sci.2010,15,229.doi:10.1016/j.cocis.2010.01.006
(45) Liggieri,L.;Miller,R.Curr.Opin.Colloid Interface Sci.2010,15,256.doi:10.1016/j.cocis.2010.02.003
(46) Huang,Y.P.;Zhang,L.;Zhang,L.;Luo,L.;Zhao,S.;Yu,J.Y.J.Phys.Chem.B2007,111,5640.doi:10.1021/jp070997t
猜你喜欢
河北水利(2022年5期)2022-06-10
哈尔滨工业大学学报(2020年1期)2020-12-21
中国生物医学工程学报(2019年6期)2019-07-16
世界农药(2019年2期)2019-07-13
纤维复合材料(2018年2期)2018-12-07
聚氯乙烯(2018年4期)2018-07-05
江西建材(2018年4期)2018-04-10
浙江大学学报(工学版)(2016年11期)2016-06-05
化工生产与技术(2016年5期)2016-03-13
火炸药学报(2014年1期)2014-03-20
