
Sulindac for stroke treatment: neuroprotective mechanism and therapy
2014-06-01 09:08JigarPravinchandraModi,HowardPrentice,Jang-YenWu
中国神经再生研究(英文版) 2014年23期
Sulindac for stroke treatment: neuroprotective mechanism and therapy
Sulindac, a widely used nonsteroidal anti-in fl ammatory drug (NSAID) is a prodrug that is reduced by methionine sulfoxide reductase to its active form as an inhibitor of cyclooxygenase 1 and 2. The drug has been shown to elicit tissue protection by processes that may include at least three functions: antioxidant, preconditioning and anti-inflammatory. Sulindac demonstrates neuroprotection that involves inhibition of mitochondrial calcium overload or a decrease in protein oxidation. We have demonstrated the induction by sulindac treatment of pro-survival proteins Hsp27, Akt and Bcl-2 in the ischemic penumbra and core of the central nervous system (CNS) infarct in a rat model of ischemic stroke. Our fi ndings point to sulindac acting on the endoplasmic reticulum (ER) to decrease ATF-6 and on the mitochondrion to increase Bcl-2 as well as decrease pro-apoptotic components BAK and PUMA. The resulting decrease in ER stress and reduction in apoptosis underlies the protective effect of sulindac in reducing infarct size following transient focal brain ischemia. The potent neuroprotective effect of sulindac in the stroke model is obtained with low-dose administration of the drug pointing to the potential of sulindac as a valuable neuroprotective agent against oxidative stress in cerebral ischemia.
Sulindac has been widely used as a NSAID that is capable of inhibiting cyclo-oxygenases (COX) 1 and 2. The molecule is a substituted indeneacetic acid chemically related to indomethacin (Strong et al., 1985). Sulindac is commercially available and known as Clinoril, a prescription NSAID that is approved for use in 34 countries (Martindales Extra Pharmacopeia), including the United States. The U.S. Food and Drug Administration have approved sulindac for treating acute gouty arthritis, acute painful shoulder (bursitis/tendonitis), osteoarthritis, and rheumatoid arthritis (http://www.fda. gov). In addition to its established anti-inflammatory, antipyretic, and analgesic properties, sulindac is thought to provide anticarcinogenic effects based on extensive data from cell culture experiments and animal model systems as well as more limited data from early-phase chemoprevention trials conducted primarily among adenomatous polyposis patients (Reid et al., 2008). Following ingestion, the parent compound (sulindac sulfoxide) is converted into sulindac sul fi de, which appears to be the primary COX-inhibiting metabolite. The second major metabolite, sulindac sulfone (exisulind), has also been shown to interrupt carcinogenesis albeit through COX-independent pathways (Brunell et al., 2011) and has undergone more limited clinical development to date in light of some toxicity concerns. The sul fi de metabolite is an active moiety and is approximately five times more potent than sulindac, while the sulfone elicits no pharmacological anti-in fl ammatory response (Strong et al., 1985). Thus sulindac may be regarded as a prodrug that is reduced by the enzyme methionine sulfoxide reductase (Msr) to the active form, with the sul fi de as the active species or pharmacophore (Moench et al., 2009). The long half life of the sul fi de results in its accumulation during chronic dosing. After a single oral dose of sulindac to man, the peak plasma concentrations of both the sul fi de and the sulfone occur about 2 hours after that of the parent compound. This suggests that the tissues rather than the gut fl ora are the site of both the reduction and oxidation of sulindac (Strong et al., 1985).
In addition to its known anti-inflammatory activity there have been numerous studies in recent years on the ability of sulindac and its metabolites to act as potential anti-cancer agents, based on their ability to slow the progression of colorectal polyps to colon cancer, as well as their ability to kill colon cancer cells and other types of cancer cell (Marchetti et al., 2009). However, the potential value of sulindac for protection in acute cerebral ischemia is still uncharacterized. A growing body of evidence shows that sulindac offers remarkable neuro-protective effects by inhibition of mitochondrial Ca2+overload or reduction of protein oxidation in neurodegenerative disorders (Xing et al., 2012) indicating that sulindac may have potential to exert a bene fi cial effect in cerebral ischemia.
Recent studies suggest that sulindac protects normal cells against oxidative damage. Previous studies on the heart suggested that sulindac protection against ischemic damage occurs through an ischemic preconditioning mechanism.
Sulindac was found to induce inducible nitric oxide synthase and Hsp 27 in a PKC dependent manner. It has been widely proposed that compounds that could precondition cells to oxidative stress may have important therapeutic value, since oxidative damage appears to play a major role in age related diseases (Moench et al., 2009).
A number of studies show ef fi cacy of sulindac in models of neurodegenerative disease and importantly the drug is capable of crossing the blood brain barrier (BBB) and therefore may be capable of interacting with therapeutic targets in the CNS. Evidence of the sulindac’s ability to cross the BBB is found in studies on guinea pig (Duggan et al., 1980). They detected signi fi cant levels of sulindac sulfoxide, sulindac sul fi de and sulindac sulfone in brain after single IV injections of C-14 labeled sulindac sulfoxide. In a study, injection with either sulindac epimer (the S-epimer or the R-epimer) in rats resulted in production of both the sulfone and sul fi de metabolites in brain (Brunell et al., 2011). Several studies point to the likelihood that sulindac may protect against the progression of Alzheimer’s disease and Parkinson’s disease either through generalized anti-in fl ammatory actions or through speci fi c effects on protein aggregate formation. Epidemiological studies have indicated a lower risk of developing Alzheimer’s disease or Parkinson’s disease associated with use of non-aspirin NSAIDs. NSAIDS have been proposed as agents that modulate Aβ production and sulindac was found to reduce levels of secreted Aβ42in cell culture and to decrease levels of soluble Aβ (1—42) in brains from the Tg2576 mutant mouse. It has been proposed recently that NSAID based γ-secretase modulators may bind directly to the APP/C99 substrate to form a complex that modulates γ-secretase cleavage. This mechanism is controversial however and in contrast it has also been suggested that monomeric γ secretase modulators either do not bind to C99 or Aβ42or only bind non-speci fi cally and weakly. While epidemiological studies on Parkinson’s disease point to a decreased incidence with NSAID use, the mechanisms of NSAID protection in Parkinson’s remains to be elucidated.The aggregation of α synuclein (αS) in the brain is a key stepin the development of Parkinson’s disease. It has been proposed that the precursors of fi brils of αS (f-αS) may be more toxic than the fi brils themselves (Hirohata et al., 2008). Using in vitro assays, Hirohata et al. (2008) has shown that sulindac inhibits formation of f-αS and destabilized preformed f-αS.
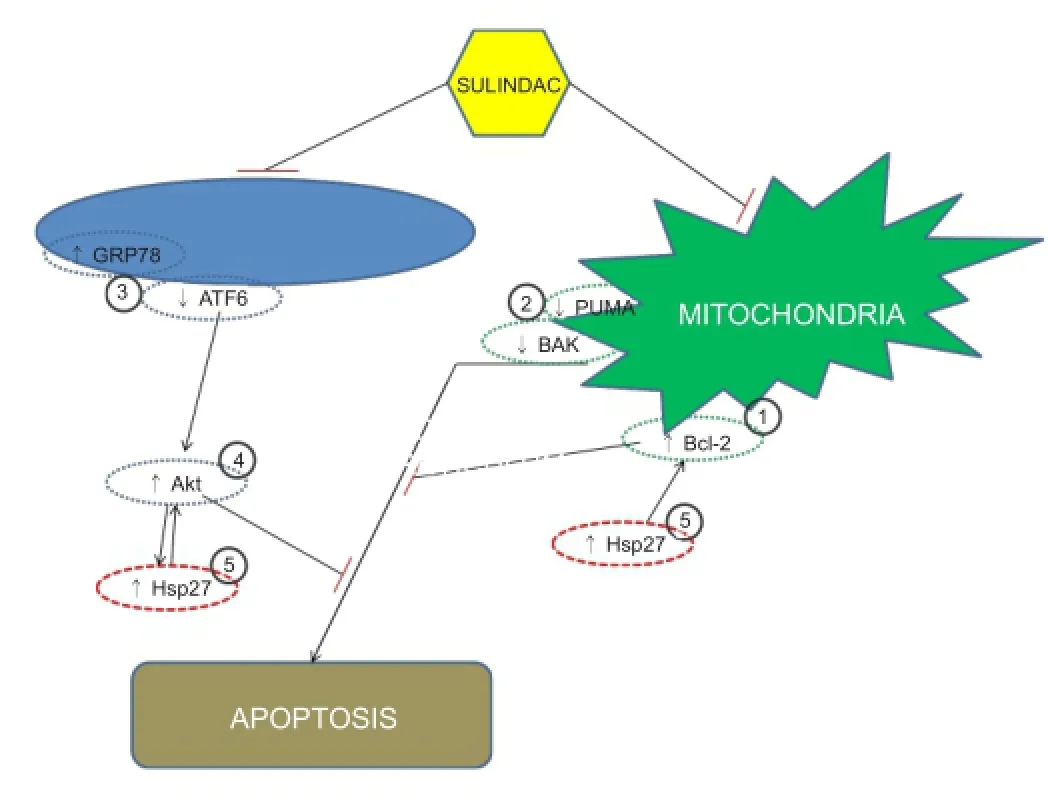
Figure 1 Proposed mechanism of action of sulindac.
We discovered that sulindac at 0.2 mg/kg reduced the brain infarct size when it was administered either before or after middle cerebral artery occlusion (MCAO) surgery. Speci fi cally, animals that were sham operated or MCAO operated with or without sulindac treatment were sacri fi ced 3 & 11 days after stroke onset, and infarct size in the left hemisphere was measured by 2, 3, 5-triphenyltetrazolium chloride (TTC) staining. Western blotting on tissue from the core and the penumbra of both hemispheres was employed for analysis of the expression of key proteins involved in apoptosis (Bcl-2 family members), and cell protection and survival (two heat shock proteins namely Hsp27 and Hsp70 as well as the signaling kinase Akt). TTC analysis of brain slices indicated a decrease in infarct size in sulindac treated animals at 4 mm, 6 mm and 8 mm from the anterior pole. The western results indicated that sulindac induced Hsp 27 protein expression in ischemic penumbra and core on days 3 & 11. Hsp 27 is a marker of cell stress and plays an important role as a molecular chaperone. There were also signi fi cant increases in the protective molecules Akt and Bcl-2 in ischemic penumbra and core (Modi et al., 2014).
Conceptually, the idea of proposing to use sulindac as a novel therapy for stroke is innovative. Despite extensive research to develop drugs for stroke based on the known mechanisms such as glutamate receptor antagonists, calcium channel blockers, enzyme inhibitors, inhibitors of apoptotic pathways and ROS scavengers, the efforts are disappointing. This is partially due to the fact that the underpinning mechanism of stroke-induced brain injury is multi-factorial, and hence it needs a therapeutic intervention that can address the complicated nature of the disease. Sulindac may act therapeutically in cerebral ischemia through its effects on Hsp 27 and Akt (Figure 1). As Hsp 27 and Akt prevent apoptosis it is likely that the elevated levels of the anti-apoptotic molecule Bcl-2 in the current study will contribute by further augmenting the pro-survival effect of sulindac. Hence sulindac exerts its neuroprotective role by inhibiting apoptosis via increased Bcl-2 expression and decreased BAK and PUMA expression in the ischemic penumbra (Figure 1).

Figure 2 Akt expression in penumbra of ischemic area of stroke on day 11 (After stroke surgery, animals were sacri fi ed on day 11).
An enhanced expression of GRP78 in the sulindac treated ischemic core and ischemic penumbra implies that sulindac may protect the brain through preconditioning and is consistent with previous studies on ischemic preconditioning showing that an enhanced level of GRP78 expression and protection against slow neuronal death (Modi et al., 2014).
In our study, Akt is activated after ischemia. The sulindac treated groups show greater than 3 fold Akt activation in the penumbra of the ischemic model of stroke compared to the penumbra of the untreated group and approximately a 50% increase in the core at day 3 after vessel occlusion (Modi et al., 2014). At day 11 after vessel occlusion, the sulindac treated group shows a greater than 2 fold increase in Akt in the penumbra compared to the untreated group and an approximately 1.5 fold increase in the penumbra after post ischemia treatment (after 24 hour stroke) with sulindac compared to the untreated group (Figure 2).
None of the currently available pharmacological interventions can provide an effective treatment for stroke. This is partially due to the fact that the underpinning mechanism of stroke-induced brain injury is multi-factorial, and hence it needs a therapeutic intervention that can address the complicated nature of the disease. Sulindac is a substrate for the enzyme methionine sulfoxide reductase which reduces sulindac to sulindac sulfide and elicits the anti-inflammatory action of the drug. A second mechanism of action of sulindac is through acting in conjunction with the catalytic antioxidant action of Msr to decrease cellular oxidant levels (Weissbach et al., 2005). A further mode of action by sulindac is as a preconditioning agent switching on key protective pathways as was previously reported in studies on myocardial ischemia. Hence, sulindac can combat types of brain injury in stroke that are either related to in fl ammation or to oxidative stress both of which are believed to be important mechanisms underlying tissue damage in stroke. Based on its mode of action as described above, the use of sulindac, a potent anti-oxidant and anti-in fl ammatory agent, in stroke interventions is both innovative and novel. Another innovative feature of the current application of sulindac is its safety and ef fi cacy since sulindac is a U.S. Food and Drug Administration approved drug and it has been widely used clinically fordecades. Furthermore, in this invention we have shown that sulindac at low concentration is effective in reducing the size of brain infarction by increasing the level of molecules that are important for cell protection and survival.
In future experiments, we plan to speci fi cally address the mechanism of protection by sulindac, and also test whether, sulindac is functioning as a preconditioning agent in stroke. To understand this protective mechanism we will evaluate changes in preconditioning markers, the role of reducing enzymes and mitochondrial function. In conclusion, we believe that sulindac may represent a novel therapeutic agent for oxidative stress induced ischemic diseases. Sulindac and its metabolites, sulindac sulfide and sulindac sulfone, inhibit the activation of the nuclear factor-kappaB (NF-κB) pathway by inhibiting IKKb kinase activity in colon cancer (Yamamoto et al., 1999). This result suggests that inhibition of components of the NF-κB pathway may at least in part be involved in the anti-inflammatory properties of sulindac. Investigation of the NF-κB pathway in ischemic brain represents an important future direction for elucidating the mechanisms of protection of sulindac in stroke.
The reported side effects of sulindac are important to consider and are dependent on dose and timing of administration. Sulindac has been reported to elicit gastrointestinal damage because of inhibition of PGE synthesis through COX inhibition. In circumstances where potential gastrointestinal de fi cits are a concern clinically, administration of the antacid lansoprazole has offered an effective route for abolishing such adverse effects. Many NSAIDs including sulindac are reported clinically to increase the risk of heart attacks (Shau et al., 2012). As a possible solution to this problem, the low dose administration of sulindac employed for tissue protection in a stroke model by Modi et al. (2014) and in a model of myocardial ischemia by Moench et al. (2009) may not elicit COX inhibition and could avoid potential cardiovascular side effects. Hypertensive side-effects may arise from the use of NSAIDs and these are likely to depend on the speci fi c NSAID used as well as the type of antihypertensive agent used if they are taken concurrently. Two meta-analyses based on data from younger adults demonstrated that NSAID use results in an increase in mean blood pressure of 5.0 mm Hg. Comparison of different NSAIDs demonstrated that piroxicam and indomethacin showed the largest and sulindac the smallest pressor effect.
The introduction of sulindac as a pharmacological intervention will greatly improve the effectiveness of stroke treatment over traditional drug treatments since it will combat two of the most likely mechanisms involved in the stroke-induced brain injury. The future clinical impact of sulindac treatment to the fi eld of therapeutic intervention for brain diseases including stroke, Parkinson’s disease and Alzheimer’s disease will be highly signi fi cant. The novel application of this drug has already been demonstrated as a “proof of concept” for treatment of stroke and other brain diseases.
The protective effect we have observed with sulindac in cerebral ischemia is contrary to several studies that NSAIDs cause increased risk of stroke and heart attack. It should be noted that in the rat feeding experiments described in this study the daily dose of sulindac (0.2 mg/d) was only 10—15%, on a weight basis, compared to the doses taken clinically as an anti-in fl ammatory agent. Hence sulindac at the low dose administered in this study is likely to be very valuable as a neuro-protective agent against oxidative stress in cerebral ischemia (Modi et al., 2014).
This study was supported in part by James & Esther King Biomedical Research Grant # 09KW-11 and Florida Atlantic University Major Research Theme in Neuroscience Grant.
Jigar Pravinchandra Modi, Howard Prentice, Jang-Yen Wu College of Medicine, Florida Atlantic University, Boca Raton, USA (Modi JP,
Prentice H, Wu JY)
Program in Integrative Biology, Florida Atlantic University
(Prentice H, Wu JY)
Centre of Complex Systems and Brain Sciences, Florida Atlantic University, USA (Modi JP, Prentice H, Wu JY)
Taichung
Brunell D, Sagher D, Kesaraju S, Brot N, Weissbach H (2011) Studies on the metabolism and biological activity of the epimers of sulindac. Drug Metab Dispos 39:1014-1021.
Duggan DE, Hooke KF, Hwang SS (1980) Kinetics of the tissue distributions of sulindac and metabolites. Relevance to sites and rates of bioactivation. Drug Metab Dispos 8:241-246.
Hirohata M, Ono K, Morinaga A, Yamada M (2008) Non-steroidal anti-in fl ammatory drugs have potent anti- fi brillogenic and fi bril-destabilizing effects for alpha-synuclein fi brils in vitro. Neuropharmacology 54:620-627.
Marchetti M, Resnick L, Gamliel E, Kesaraju S, Weissbach H, Binninger D (2009) Sulindac enhances the killing of cancer cells exposed to oxidative stress. PLoS One 4:e5804.
Modi JP, Gharibani PM, Ma Z, Tao R, Menzie J, Prentice H, Wu JY (2014) Protective mechanism of sulindac in an animal model of ischemic stroke. Brain Res 1576:91-99.
Moench I, Prentice H, Rickaway Z, Weissbach H (2009) Sulindac confers high level ischemic protection to the heart through late preconditioning mechanisms. Proc Natl Acad Sci U S A 106:19611-19616.
Reid JM, Mandrekar SJ, Carlson EC, Harmsen WS, Green EM, McGovern RM, Szabo E, Ames MM, Boring D, Limburg PJ (2008) Comparative bioavailability of sulindac in capsule and tablet formulations. Cancer Epidemiol Biomarkers Prev 17:674-679.
Shau WY, Chen HC, Chen ST, Chou HW, Chang CH, Kuo CW, Lai MS (2012) Risk of new acute myocardial infarction hospitalization associated with use of oral and parenteral non-steroidal anti-in fl ammation drugs (NSAIDs): a case-crossover study of Taiwan’s National Health Insurance claims database and review of current evidence. BMC Cardiovasc Disord 12:4.
Strong HA, Warner NJ, Renwick AG, George CF (1985) Sulindac metabolism: the importance of an intact colon. Clin Pharmacol Ther 38:387-393.
Weissbach H, Resnick L, Brot N (2005) Methionine sulfoxide reductases: history and cellular role in protecting against oxidative damage. Biochim Biophys Acta 1703:203-212.
Xing Y, Zhang X, Zhao K, Cui L, Wang L, Dong L, Li Y, Liu Z, Wang C, Zhang X, Zhu C, Qiao H, Ji Y, Cao X (2012) Bene fi cial effects of sulindac in focal cerebral ischemia: a positive role in Wnt/β-catenin pathway. Brain Res 1482:71-80.
Yamamoto Y, Yin MJ, Lin KM, Gaynor RB (1999) Sulindac inhibits activation of the NF- B pathway. J Biol Chem 274:27307-27314.
Jang-Yen Wu, Ph.D.; Howard Prentice, Ph.D.
Email: jwu@fau.edu; hprentic@fau.edu.
10.4103/1673-5374.147919 http://www.nrronline.org/
Accepted: 2014-11-29
Modi JP, Prentice H, Wu JY. Sulindac for stroke treatment: neuroprotective mechanism and therapy. Neural Regen Res. 2014;9(23):2023-2025.

- 中国神经再生研究(英文版)的其它文章
- “Standby” EMT and “immune cell trapping” structure as novel mechanisms for limiting neuronal damage after CNS injury
- Angioplasty and stenting for severe vertebral artery ori fi ce stenosis: effects on cerebellar function remodeling veri fi ed by blood oxygen level-dependent functional magnetic resonance imaging
- Lesion localization of global aphasia without hemiparesis by overlapping of the brain magnetic resonance images
- A novel functional electrical stimulation-control system for restoring motor function of post-stroke hemiplegic patients
- Neurotrophins and their receptors in satellite glial cells following nerve injury
- Dynamic reactive astrocytes after focal ischemia