
RISK途径在二氮嗪后处理离体大鼠心肌中的保护作用
2014-05-18 08:09刘兴奎
中国药理学通报 2014年9期
王 英,谢 平,张 琳,刘兴奎,喻 田
(1.青岛市市立医院麻醉科,山东青岛 266011;2.贵州省麻醉与器官保护基础研究重点实验室,贵州遵义 563003)
磷酯酰肌醇3-激酶-蛋白激酶B(phosphatidy qinositol 3-kinase-protein kinase B,PI3K/Akt)途径和细胞外调节激酶(extracellular signal-regulated kinase 1/2,ERK1/2)途径共同组成了再灌注损伤挽救激酶(reperfusion injury salvage kinase,RISK)途径,在心肌缺血/再灌注损伤中起重要的保护作用。目前,在离体动物的实验中发现钾通道开放剂(potassium channel openers,KCOs)的心肌保护作用是通过胞膜KATP通道(sarcolemmal ATP sensitive potassium channel,sarc-KATP)和线粒体 KATP通道(mitochondrial ATP sensitive potassium channel,mito-KATP)而实现的,并认为后者起主要作用。然而,在特异性mito-KATP开放剂二氮嗪(diazoxide,DZ)后处理减轻离体大鼠心肌缺血/再灌注损伤中,是否通过RISK途径而起到保护作用还鲜有报道。本研究拟采用Langendorff离体心脏灌注模型,应用DZ行药物后处理,观察其对缺血/再灌注心脏功能的影响,并探讨DZ后处理是否通过激活RISK信号通路减轻离体大鼠缺血/再灌注损伤。
1 材料
1.1 实验动物及分组 健康Sprague-Dawley成年大鼠32只(♀♂各半,由第三军医大学大坪医院实验动物中心提供,清洁级,证书号:SCXK(军)2002008,随机分为正常组(NOR)、对照组(CON)、二氮嗪后处理组(DZ)、LY拮抗二氮嗪组(DZ+LY)。
1.2 主要试剂及仪器 Akt、Phospho-Akt、p70S6、Phospho-p70S6、eNOS、Phospho-eNOS、p44/42 MAP Kinase、Phospho-p44/42 MAP Kinase兔抗大鼠多克隆抗体(均为 CST公司产品),辣根过氧化物酶(HRP)标记山羊抗兔 IgG(DakoCytomation公司),预染蛋白分子marker(Biolabs公司),BCA蛋白浓度测定试剂盒(江苏碧云天生物技术研究所),二氮嗪(diazoxide)、LY294002(LY)、二甲基亚砜(DMSO)、甲叉双丙烯酰胺(bis-Acr)、三羟甲基氨基甲烷(hydroxymethyl aminomethane,Tris)、N-2-羟-酸 哌嗪-N′-2-乙磺酸(HEPES)均为 Sigma公司,其他生化试剂为国产分析纯;Langendorff离体心脏灌注模型(西班牙,PanLab公司)、Powerlab/8SP实验数据采集系统(澳大利亚,ADInstrument公司)、台式高速冷冻离心机(Eppendorf,5804R型)、-80℃低温冰箱(Forma,美国)、ELX-800酶标仪(BIO-TEC.INC,美国)、Mini Trans-Bloe电泳仪(BIO-RAD,美国)、全自动凝胶图像分析仪(美国Syngene公司)。
1.3 主要液体配制 ①Kerbs-Henseleit缓冲液(成分单位:mmol· L-1,NaCl:118.00,KCl:4.75,CaCl2:2.50,NaHCO3:24.80,MgCl26H2O:1.19,KH2PO4:1.19,glucose:11.1);ST.Thomas停跳液(成分单位:mmol·L-1,NaCl:110.00,KCl:16.00,CaCl2:1.20,MgCl2:16.00,NaHCO3:10.00 pH 7.8);②8%SDS-PAGE分离胶30%丙烯酰胺、1.5 mol·L-1Tris缓冲液(pH 8.8)、10%APS、10%SDS、TEMED及5%SDS-PAGE积层胶;③ 电泳液(25 mmol·L-1Tris、250 mmol·L-1甘氨酸、0.1%SD)及电转缓冲液(25 mmol·L-1Tris base、0.2 mol·L-1甘氨酸、20%甲醇);④ PBS-T缓冲液 0.01 mol·L-1磷酸盐缓冲液加0.1%Tween-20。
2 方法
2.1 离体大鼠Langendroff全心缺血/再灌注模型的建立 大鼠ip给予戊巴比妥钠40 mg·kg-1麻醉,肝素钠500 U·kg-1抗凝,迅速开胸取心脏,立即放入预冷的K-H液(4℃),轻轻挤压心脏洗净残留血液,液面下主动脉插管,固定置于Langendorff灌注管口。用37℃预先氧平衡的K-H液5.80kPa心脏逆行灌注,心脏跳动后于左心耳剪一小口,将带有乳胶水囊的测压管经二尖瓣插入左心室,连接Powerlab/8SP生物机能试验压力换能器系统,调节水囊使LVEDP为0.93kPa。
2.2 灌注方案 离体心脏37℃的K-H液平衡灌注20 min,平衡后监测 HR>200次/分、LVSP>10kPa、室性早搏<2个/分,未达条件者舍弃。NOR组平衡后无处理续灌70 min;CON组平衡后,缺血40 min,再续灌30 min;DZ组于再灌注开始即刻,主动脉逆灌50μmol·L-1二氮嗪5min;DZ+LY组于再灌注开始即刻主动脉灌注LY 15μmol·L-1持续5 min后,二氮嗪后处理5 min;除去NOR组均给予灌注4℃ST.Thomas停跳液10 ml·kg-1,继之停止灌注泵造成全心缺血,所有心脏在常温下缺血40 min后,再次灌注37℃含氧灌注液30 min后,取左室组织迅速置于液氮中保存,分别于平衡20 min末和再灌注30 min末时采集心脏功能各项参数。
2.3 心脏功能测定 各组分别于平衡末和再灌注30 min末测定下列心脏血液动力学参数:心率(heart rate,HR)、冠脉流量(coronary flow,CF)、左心室发展压(left ventricular developed pressure,LVDP)、左心室舒张末压(left ventricular end-diastolic pressure,LVEDP)、左心室内压上升最大速率(themaximum rate of increase of left ventricular pressure,+d p/d tmax)和左心室内压下降最大速率(the maximum rate of decrease of left ventricular pressure,-d p/d tmax)。
2.4 蛋白质的提取及W estern blot检测心肌Akt、P70S6K、eNOS、ERK磷酸化水平 从液氮中取出心肌组织称量,按3 ml·g-1比例加入4℃组织裂解液(50mmol·L-1Tris.HCl、pH 7.4、1 mmol·L-1PMSF、50 mmol·L-1NaF、150 mmol·L-1NaCl、5 mmol·L-1EDTA、0.45mmol·L-1EGTA、1%Triton X-100、2 m l·g-1aprotinin、2 m l·g-1leupeptin、5 mg·L-1pepstatin、1%Tween-20),冰浴中剪碎组织,超声匀浆至组织成糜状,静置1 h,14 000×g离心1 h,取上清,14 000×g再离心30 min,取出上清液,用BCA法测定待测样品的蛋白浓度(按BCA蛋白浓度检测试剂盒说明书操作),最后取上清蛋白分装。分别根据目的蛋白分子量配制8%SDS-PAGE分离胶,上样量为20μg/20μl,按标本与上样缓冲液体积4∶1加入5X上样缓冲液,总体积25μl。混匀后煮沸5 min,上样电泳、转膜,一抗孵育过夜,二抗标记孵育1 h,荧光显色剂染膜,X线片曝光,显影、定影。采用Image-ProPlus图像分析软件(Version4.1,Media Cybernetics,LP,USA)对蛋白条带进行半定量分析。
2.5 统计学处理 实验数据按完全随机对照设计的要求收集整理,所有数据用¯x±s表示,各组间数据计量资料用SPSS13.0统计软件进行单因素方差(ANOVA)分析,处理前后数据用配伍t检验。
3 结果
3.1 离体大鼠心脏心功能 4组离体鼠心平衡末、再灌注末30min时心率(HR)、冠脉流量(CF)、左心室发展压(LVDP)、左心室舒张末压(LVEDP)、压力瞬时最大变化率(±d p/d tmax)结果如下(Tab 1~3)。各组间平衡末比较差异无显著性(P>0.05);NOR组组内再灌注30 min与平衡末比CF差异无显著性,余各组组内再灌注30 min与平衡末比 HR、LVDP、±d p/d tmax明显降低(P<0.01),LVEDP明显增加(P<0.01);DZ组再灌注末 HR、CF、LVDP、±d p/d tmax与CON组、DZ+LY组比明显增加(P<0.01),LVEDP明显降低(P<0.01)。
Tab 1 Change of isolated heart HR and CF in four groups(±s,n=8)
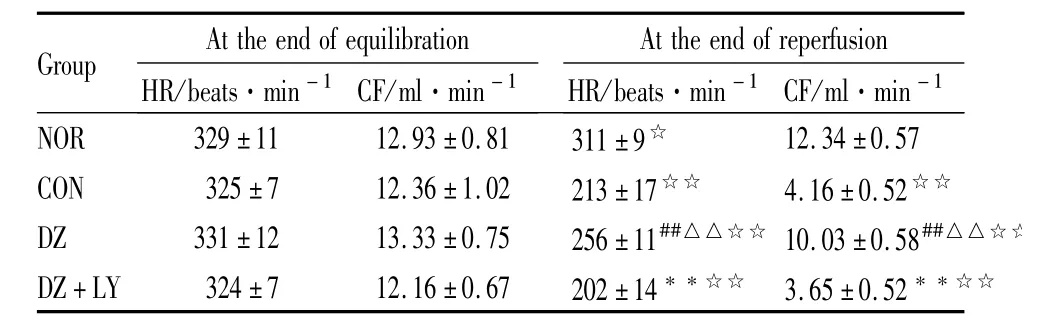
Tab 1 Change of isolated heart HR and CF in four groups(±s,n=8)
##P<0.01 vs NOR;△△P<0.01 vs CON;**P<0.01 vs DZ;☆P<0.05,☆☆P<0.01 30min-reperfusion vs20min-equilibration.
Group At theend ofequilibration HR/beats·min-1 CF/ml·min-1 At theend of reperfusion HR/beats·min-1 CF/ml·min-1 NOR 329±11 12.93±0.81 311±9☆12.34±0.57 CON 325±7 12.36±1.02 213±17☆☆ 4.16±0.52☆☆DZ 331±12 13.33±0.75 256±11##△△☆☆ 10.03±0.58##△△☆☆DZ+LY 324±7 12.16±0.67 202±14**☆☆ 3.65±0.52**☆☆
Tab 2 Change of isolate hearts LVDP and LVEDP in four groups(x¯±s,n=8)
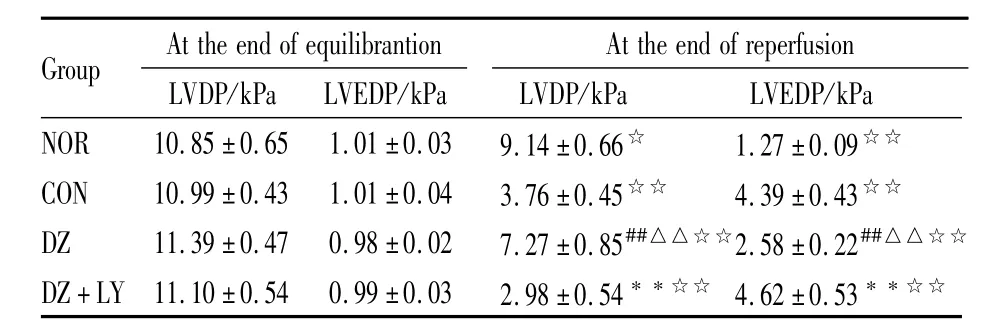
Tab 2 Change of isolate hearts LVDP and LVEDP in four groups(x¯±s,n=8)
##P<0.01 vs NOR;△△P<0.01 vs CON;**P<0.01 vs DZ;☆P<0.05,☆☆P<0.01 30min-reperfusion vs20min-equilibration.
Group At the end ofequilibrantion At the end of reperfusion LVDP/kPa LVEDP/kPa NOR 10.85±0.65 1.01±0.03 9.14±0.66☆ 1.27±0.09 LVDP/kPa LVEDP/kPa☆☆CON 10.99±0.43 1.01±0.04 3.76±0.45☆☆ 4.39±0.43☆☆DZ 11.39±0.47 0.98±0.02 7.27±0.85##△△☆☆2.58±0.22##△△☆☆DZ+LY 11.10±0.54 0.99±0.03 2.98±0.54**☆☆ 4.62±0.53**☆☆
Tab 3 Change of isolate hearts+d p/d t m ax and-d p/d t m ax in four groups¯x±s,n=8)

Tab 3 Change of isolate hearts+d p/d t m ax and-d p/d t m ax in four groups¯x±s,n=8)
##P<0.01 vs NOR;△△P<0.01 vs CON;**P<0.01 vs DZ;☆☆P<0.01 30min-reperfusion vs 20min-equilibration
Group-1 NOR 255±18 212±16 225±13☆☆ 172±12 At theend ofequilibrantion+d p/d t max/kPa·s-1-d p/d t max/kPa·s-1 At theendof reperfusion+d p/d t max/kPa·s-1-d p/d t max/kPa·s☆☆CON 263±16 205±22 115±21☆☆ 47±8☆☆DZ 273±18 235±17 165±12##△△☆☆ 113±11##△△☆☆DZ+LY 257±18 209±13 91±21**☆☆ 46±12**☆☆
3.2 W estern blot检测心肌 Akt、P70S6K、eNOS、ERK磷酸化水平 再灌注末DZ组Akt、P70S6K、eNOS磷酸化水平明显高于NOR组、CON组、DZ+LY组(P<0.01),但各组ERK的磷酸化水平没有明显区别,p-Akt/t-Akt、p-P70S6/t-P70S6、p-eNOS/t-NOS、p-ERK/t-ERK为蛋白条带灰度值的比值,见Fig 1~8。
3.2.1 Akt磷酸化表达情况 见Fig 1、2。
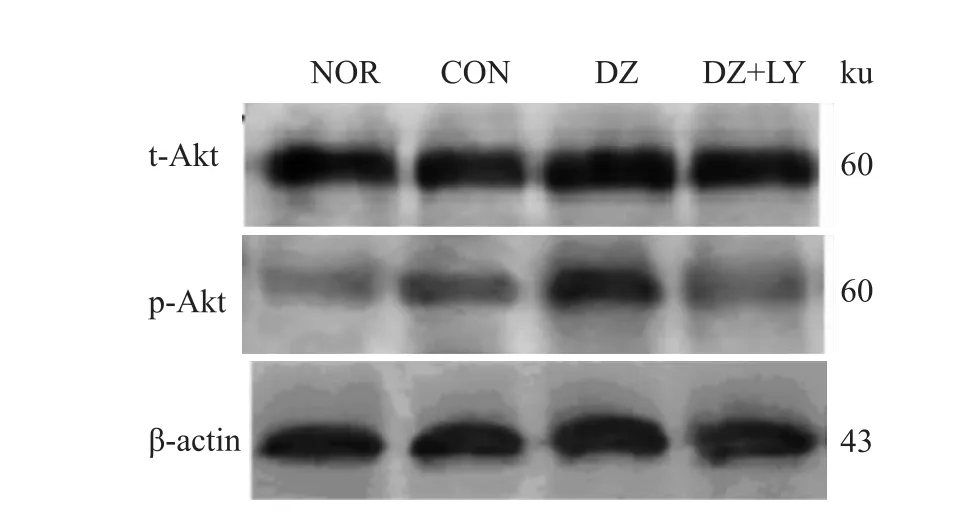
Fig 1 Four groupsof total Akt and phosphorylated Akt electrophoresis images
3.2.2 P70S6磷酸化表达情况 见Fig 3、4。
3.2.3 eNOS磷酸化表达情况 见Fig 5、6。
3.2.4 ERK磷酸化表达情况 见Fig 7、8。

Fig 2 Level of phosphoric acid expressions of Akt in LV of rats from four groups(¯x±s,n=3)##P<0.01 vs NOR;△△P<0.01 vs CON;**P<0.01 vs DZ
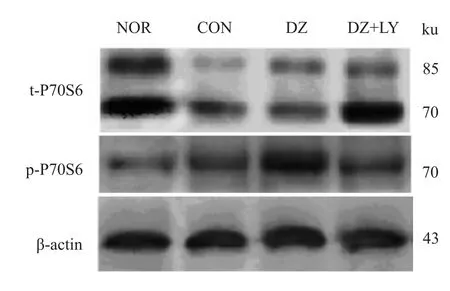
Fig 3 Four groups of total P70S6 and phosphorylated P70S6 electrophoresis images
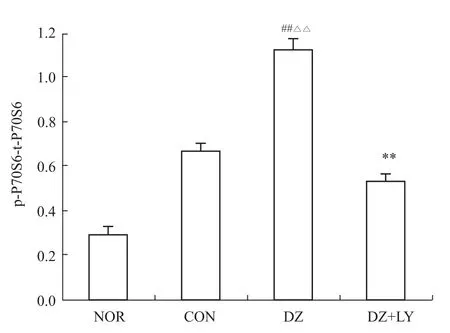
Fig 4 Level of phosphoric acid expressions ofP70S6 in LV of rats from four groups(±s,n=3)##P<0.01 vs NOR;△△P<0.01 vs CON;**P<0.01 vs DZ
4 讨论
心肌缺血/再灌注损伤(ischemia reperfusion injury,IRI)是临床上常见的病理生理过程,人们对IRI的机制和抗损伤的措施进行了大量研究,其中2003年Zhao等[1]提出了缺血后处理(ischemicpostconditioning,IPTC),并在动物模型上证实了后处理的保护效果,IPTC能够有效地减轻再灌注损伤,而药物后处理是在缺血后再灌注之前给予某种药物使之产生与IPTC相似的保护效果。目前研究表明,二氮嗪、挥发性麻醉药、胰岛素、缓激肽等药物都具有后处理的保护作用[2-4]。二氮嗪[5]、七氟醚[6]及多种细胞因子均可通过激活PI3K-Akt信号转导系统,抑制心肌细胞凋亡,促进心肌细胞存活,其中Akt活化是这些信号转导通路发挥心脏保护作用的关键。目前PI3K/Akt和ERK1/2作为治疗新靶点日益受到关注。具体的说,IPTC激活RISK途径,其上游可能是激活了G蛋白偶联受体如腺苷受体,尤其是A2和A3受体,其下游可能激活了GSK3β、eNOS、蛋白激酶Cε(PKCε),最终作用于线粒体和钾离子通道,减少mPTP开放和增加钾离子通道开放[7-9],从而保护心肌细胞抵抗细胞内和线粒体内钙超载、氧化应激反应和ATP耗竭。
本研究结果表明,与CON组和DZ+LY组比,DZ后处理组,其 LVDP、HR、CF、+d p/d tmax、-d p/d tmax明显增加(P<0.01),LVEDP明显降低(P<0.01),能明显改善离体缺血/再灌注大鼠心脏的功能,提示DZ后处理能有效减轻IRI,具有心肌保护作用,表明mito-KATP敏感性钾通道在后处理中起到关键作用。本实验中,再灌注末DZ组 PKB/Akt、eNOS、P70S6K磷酸化水平的表达明显高于 NOR组、CON组,而PI3K抑制剂LY294002可取消这种作用,并取消DZ改善心功能的效应。各组ERK1/2磷酸化水平的表达差异无统计学意义(P>0.05)。表明二氮嗪后处理可能激活了PI3K-Akt信号途径,并且也激活其下游靶点eNOS、P70S6K,使它们磷酸化水平增强,从而抑制心肌细胞的凋亡,产生心肌保护效应。但各组ERK的磷酸化水平没有明显区别,提示DZ后处理并没有影响ERK1/2磷酸化的表达。

Fig 5 Four groups of total eNOS and phosphorylated eNOS electrophoresis images
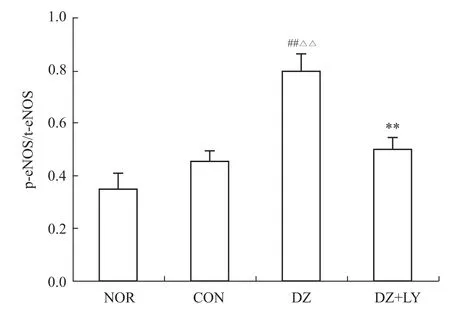
Fig 6 Level of phosphoric acid expressions of eNOS in LV of rats from four groups(±s,n=3)##P<0.01 vs NOR;△△P<0.01 vs CON;**P<0.01 vs DZ
Fig 7 Four groups of total ERK and phosphorylated ERK electrophoresis images
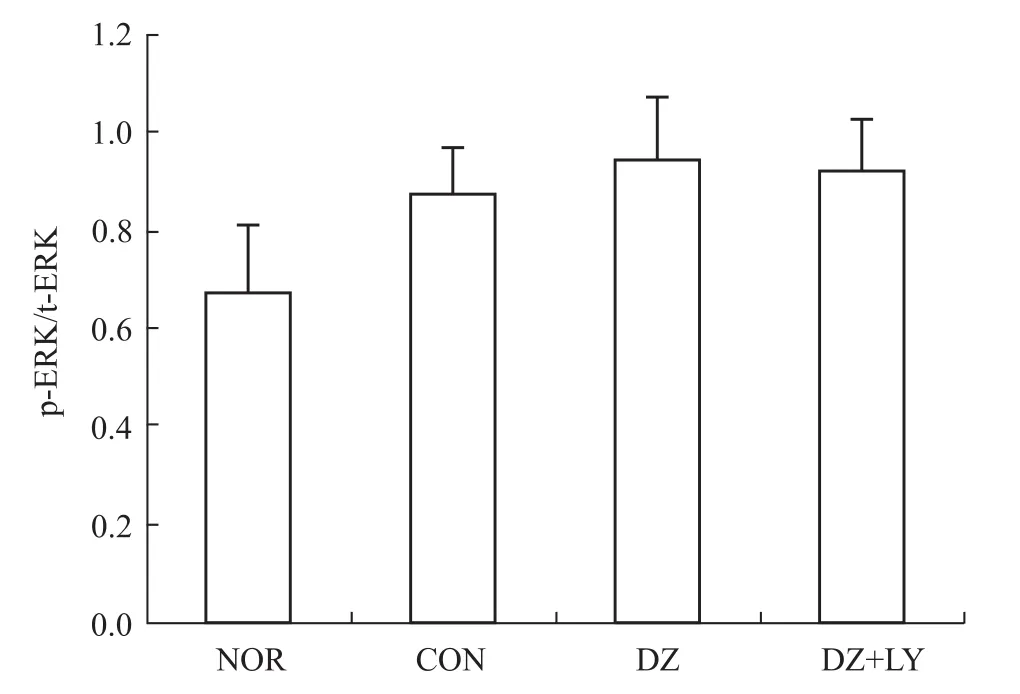
Fig 8 Level of phosphoric acid expressions of ERK in LV of rats from four groups(±s,n=3)
目前对于PI3K/Akt和ERK1/2在后处理中的相对重要性仍存有争议。Darling等[10]在兔离体心脏缺血后处理模型的研究中发现后处理激活的是ERK1/2,而非PI3K/Akt。而另一项关于缓激肽药理后处理的研究则表明ERK1/2就是PI3K/Akt的一个下游底物[11]。这些观点的分歧可能与实验动物、后处理的方式以及所取标本的部位不同有关。在以往的研究中[12]也显示,PI3K/Akt和 ERK1/2的活化无论是在心肌肥厚还是在缺血或衰竭的心脏中,都是一个复杂的多相的时间点过程。虽然本实验表明ERK1/2没有被DZ后处理所激活,但是不能排除ERK1/2在后处理中的心肌保护作用,因为我们不能排除可能积累的磷酸化在特定的亚细胞上起作用,而且蛋白质磷酸化与非磷酸化的过程本身就是个可逆的过程,在实验中也不能确定磷酸化只在单一的时间点内发生,所以对于DZ后处理是否激活ERK1/2途径,其机制如何还有待进一步研究。
本研究还发现,各组 PKB/Akt、P70S6K、eNOS磷酸化表达的变化与其对应的离体鼠心心脏功能的改变,具有一定的联系,推测DZ后处理可能是通过开放 mito-KATP通道,启动某种内源性机制激活PI3K-Akt信号途径,抑制心肌细胞的凋亡而保护心肌。因此,可以认为DZ后处理可激活PI3K-Akt信号途径,而激活的PI3K-Akt信号途径又能促使mito-KATP通道的开放,二者都能抑制心肌细胞的凋亡,协同发挥作用,最终产生心肌保护效应。
参考文献:
[1] Zhao ZQ,Corvera JS,Halkos M E,etal.Inhabition ofmyocardial injury by ischemic postconditioning during reperfusion:comparision with preconditioning[J].AM J Physiol Heart Circ Physiol,2003,285(2):579-88.
[2] 段忠心,刘兴奎,喻 田.二氮嗪后处理对大鼠离体心脏缺血/再灌注损伤的影响[J].中华麻醉学杂志,2010,30(10):1163-7.
[2] Duan Z X,Liu X K,Yu T.Effects of diazoxide postconditioning on myocardial ischemia-reperfusion injury in isolated rat hearts[J].Chin JAnesthesiol,2010,30(10):1163-7.
[3] Meybohm P,Gruenewald M,Albrecht M,et al.Pharmacological postconditioning with sevoflurane after cardiopulmonary resuscitation reducesmyocardial dysfunction[J].Crit Care,2011,15(5):241-6.
[4] Burley D S,Baxter G F.Pharmacological targets revealed by myocardial postconditioning[J].Curr Opin Pharmacol,2009,9(2):177-88.
[5] 赵其宏,张 颖,梁启胜,等.二氮嗪后处理对缺血/再灌注心肌的保护作用及其与PI3K/Akt信号通路的关系[J].中国药理学通报,2012;28(1):127-31.
[5] Zhao Q H,Zhang Y,Liang Q S,et al.Protective effect againstmyocardial ischemia/reperfusion induced by diazoxide-postconditioning and its interaction with PI3K/Akt signaling pathway[J].Chin JAnesthesiol,2012,28(1):127-31.
[6] Ma L L,Zhang F J,Kong F J,et al.Hypertrophied myocardium is refractory to sevoflurane-induced protection with alteration of reperfusion injury salvage kinase/glycogen synthase kinase 3βsignals[J].Shock,2013,40(3):217-21.
[7] Ma X J,Yin H J,Guo C Y,et al.Ischemic postconditioning through percutaneous transluminal coronary angioplasty in pigs:roles of PI3K activation[J].Coron Artery Dis,2012,23(4):245-50.
[8] Yao SY,Nataran C,Sriram S.nNOSmediated mitochondrial injury in LPS stimulated oligodendrocytes[J].Mitochondrion,2012,12(2):336-44.
[9] Zatta A J,Kin H,Lee G,et al.Infarct sparing effect ofmyocardial postconditioning is dependent on protein kinase C signalling[J].Cardiovasc Res,2006,70(2):315-24.
[10]Darling C E,Jiang R,Maynard M,et al.Postconditioning via stuttering reperfusion limits myocardial infarct size in rabbit hearts:role of ERK1/2[J].Am J Physiol Heart Circ Physiol,2005,289(4):1618-26.
[11]Yang X M,Krieg T,Cui L,et al.NECA and bradykinin at reperfusion reduce infarction in rabbitheartsby signaling through PI3K,ERK,and NO[J].JMol Cell Cardiol,2004,36(3):411-21.
[12]Rothermel B A,Berenji K,Tannous P,et al.Differential activation of stress-response signaling in load-induced cardiac hypertrophy and failure[J].Physiol Genomics,2005,23(1):18-27.
猜你喜欢
汽车实用技术(2022年15期)2022-08-19
中国科技纵横(2021年24期)2021-03-02
今日农业(2020年20期)2020-12-15
蚕桑通报(2020年1期)2020-07-10
能源(2018年10期)2018-12-08
中国土壤与肥料(2018年5期)2018-11-05
中成药(2018年6期)2018-07-11
奥秘(2016年10期)2016-12-17
中国卫生标准管理(2015年6期)2016-01-14
园艺与种苗(2015年10期)2015-02-27
