
基于自然原子轨道电荷的取代苯甲酸 pKa值预测
2014-04-12 12:05刘恩榕张扬斌钟爱国
当代化工 2014年3期
刘恩榕,张扬斌 ,钟爱国
(台州学院医药化工学院,浙江 台州 316000)
基于自然原子轨道电荷的取代苯甲酸 pKa值预测
刘恩榕,张扬斌 ,钟爱国
(台州学院医药化工学院,浙江 台州 316000)
采用密度泛函理论(DFT)和 B3LYP/3-21+G(d)基组,优化了 21 种单取代苯甲酸分子结构,发现羧基上氧原子的自然原子轨道电荷 (NBO-O) 值与其实验 pKa值之间存在良好的线性关系(R=-0.973 6),比其原子核静电势电荷 (ESP-O) 值拟合的要好。计算了 20 种典型未知 pKa值的单和多取代苯甲酸化合物的 NBO参数,代入拟合出的优势线性参数方程, 发现其预测值与流行软件 ACD Lab 6.0 预测得到的单和多取代苯甲酸的 pKa值非常接近,最大偏差 ΔpKa小于 ± 0.03, 且新方法可以估测到 pKa值小数点后 3 位数。
密度泛函理论方法;取代苯甲酸;自然原子轨道电荷;pKa
苯甲酸在食品工业和日常生活中得到了极大的应用,是一种重要的酸型消毒防腐剂[1]。其防腐(杀菌)性能与其酸碱性(pKa值)密切相关。在强酸性条件下,苯甲酸对霉菌、酵母和细菌等均有较强的抑制作用。但苯甲酸本体 p H 值较低,其抑制效果不佳,如 p H 3.5 时,0.125 %的浓度在 1 h 内可杀灭葡萄球菌,但在碱性环境下其作用就会减弱很多。苯甲酸是一比较简单的芳香酸,能发生取代的位置也较多,因此本文以取代苯甲酸类化合物为对象,通过取代苯环上的 H 原 子来调节其 p Ka值。通常它们的值可以通过实验来测定。但实验测定方法会有一定的局限性,在实际使用中,由于分子受稳定性等诸多因素的制约,使从实验上很难准确测定一些分子的 p Ka值。因此从理论和计算上寻找有效和可靠的预测酸碱性的方法成为了目前十分活跃的一个课题。由于苯甲酸类化合物还是易挥发的,因此通过分子的结构-活性关系(QSAR)方法,如应用量化参数[2]、CoMFA 法[3]、构建拓扑指数法[4]、Hammett常数法[5]、密度泛函活性理论预测法[6]等,来对多取代苯甲酸类化合物 pKa值进行预测研究(见图 1),建立具有 pKa预测能力的较佳数学模型,这对于了解苯甲酸类化合物药理及杀菌性质具有重要意义。
图 1 取代苯甲酸的分子结构?式及编号Fig.1 The structural formula of benzoic acid
1 计算方法
1.1 量化参数选取
使用 Gaussion 09W 软件,构建 21 个已知实验pKa值的取代苯甲酸的分子结构模型,运用 GaussionView 软件在 DFT B3LYP/6 -31G + ( d ), pop = ( nbo , chelpg )水平和条件下,对 21 种取代苯甲酸类化合物进行构型全优化,分别记录苯甲酸及其取代苯甲酸羧基上 O 和 H 原子上的静电势电荷(ESP)以及其自然原子轨道电荷(NBO)值。本文用 Gaussian 09W 计算,用 Gaussview 5.08 作 ESP 图和 NBO 图。当电荷数值不标出单位时,默认为电子电量。
用 ESP、NBO 电荷值(横坐标)分别对 pKa实验值(纵坐标)做图,线性拟合得到两个线性回归方程,并得到相关系数 R 。通过比较两者的相关性,即 R 值越接近-1 或 1,相关性越好,反之,R 值越接近 0,则相关性越差,由此选出拟合效果较好的量化参数方程。
1.2 回归方程检验
选取 20 种典型的未知 pKb值的,含有不同取代基的多取代苯甲酸类衍生物,用 Gaussion 09W 软件,再次优化其结构,计算其羧基 O 原子的 NBO-O 值,并将得到的 O 原子的 NBO 值代入拟合得到的线性方程,计算未知多取代苯甲酸衍生物其 pKa值,然后与最流行的 ACD-Labs 6.0 软件直接预测 pKa值进行比较,以此来检验该线性回归方程的合理性和精确度。
2 结果与讨论
2.1 羧基上 O 原子的 ESP/NBO 电荷值与其实验 pKa
计算结果见表 1 所示。对 21 种已知 pKa实验值的取代苯甲酸,我们在其羧基上 O 原子的 ESP/ NBO 参数与其已知实验 pKa值之间做两个关系图(见图 2 和图 3 所示)。由图 2 和图 3 比较可知:O原子上的 ESP、NBO 电荷值与 pKa实验值均呈负相关,相关系数 R 分别为-0.899 8,-0.973 6,相对标准偏差(SD)分别为 0.265 4,0.134 4。
通过比较羧基 O原子的自然原子轨道电荷值(NBO-O) 所得相关方程为:Y = -14.254 3-24.862 8 ×NBO-O,其相关系数 R 最大(-0.973 6),我们可以直观地发现,NBO-O 与其实验 pKa的相关性好于ESP-O 与其实验 pKa的相关性(Y= -0.181 2-6.439 7 ×ESP-O)。
2.2 羧基上 H 原子的 ESP/NBO 电荷值与其实验 pKa
为了获得更好的量子化学描述符,我们还运用Gaussion View 5.08 软件,在 DFT B3LYP/6-31G+ ( d ),pop = (nbo, chelpg) 水平和条件下, 对 21 种取代苯甲酸类化合物进行构型全优化,得到H原子的ESP-H 值和 NBO-H 值,其结果一并整理在表 1 中。用表 1 的数据(第 4 列和第 5 列数据做横坐标,对其实验 pKa值作图),在 origin 中作图 1,可以得到另外两图(未显示)。根据 origin 中作图结果,得到的回归线方程为 Y = -2.650 2 + 14.638 5×ESP-H ,相关系数 R = 0.7654 1,相对标准偏差 SD=0.384 5。由此我们可以发现 H 原子 ESP 电荷与 pKa值具有一定的相关性的,并且是正相关。
根据 origin 中作图 2,得到回归线方程为:

相关系数 R = 0.386 2,SD = 0.551 2。
由此可以发现, H 原子 ESP-H 电荷与 pKa值的相关性并不是很好;即使用 H 的 NBO-H 来预测pKa值,也不是最佳的量子化学描述符选择。
因此,我们将采用其 NBO-O 与 pKa作图所得的线性回归方程(即 Y = -14.2543 -24.8626×NBO-O),来预测未知多取代苯甲酸衍生物的 pKa值。
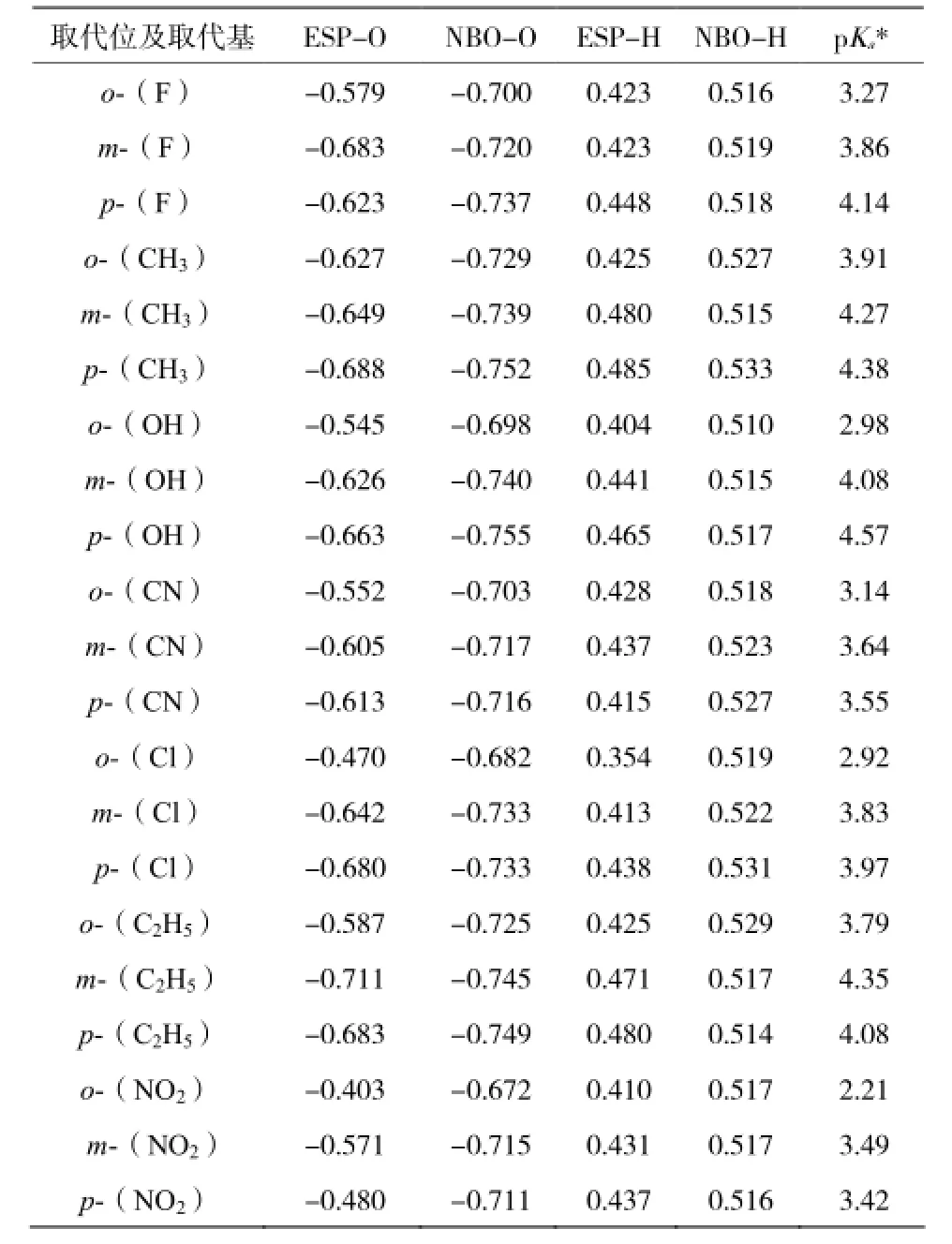
表 1 21 种单取代苯甲酸的 O(H)电荷值与其实验 pKaTable 1 The correlation charge of O(H) atom and their pKa
2.3 预测 20 种未知多取代苯甲酸的 pKa值
用如图 3 所得的较佳回归线性方程,预测了 20种未知多取代苯甲酸的 pKa值, 并与业界最流行软件 ACD-Labs 6.0 预测的 pKa值进行了比较(数据见表 2 所示)。通过比较发现,两种方法求得的 pKa的差值在允许范围内波动。两者预测结果非常接近,最大 ΔpKa< ± 0.03。而我们的新方法却可以精确到小数点后3位。
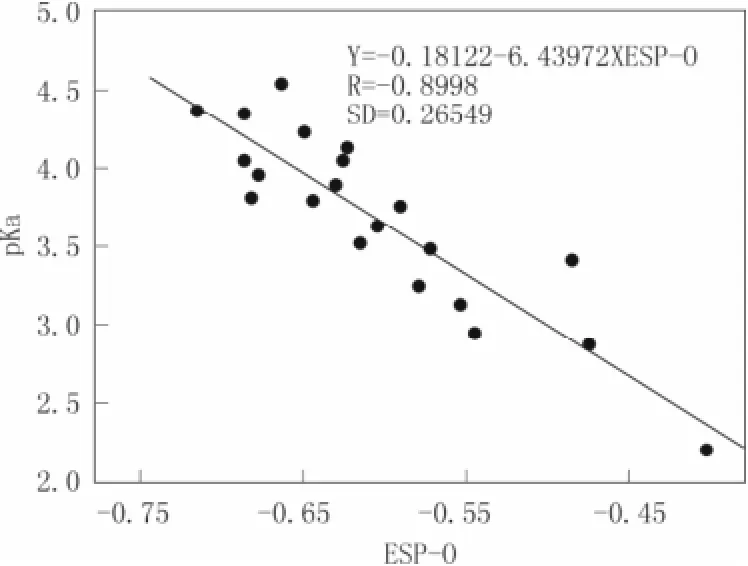
图 2 羧基上 O 原子 ESP 电荷与实验 pKa之间的关系Fig.2 The relation of experiment pKawith the ESP charges of the O atom
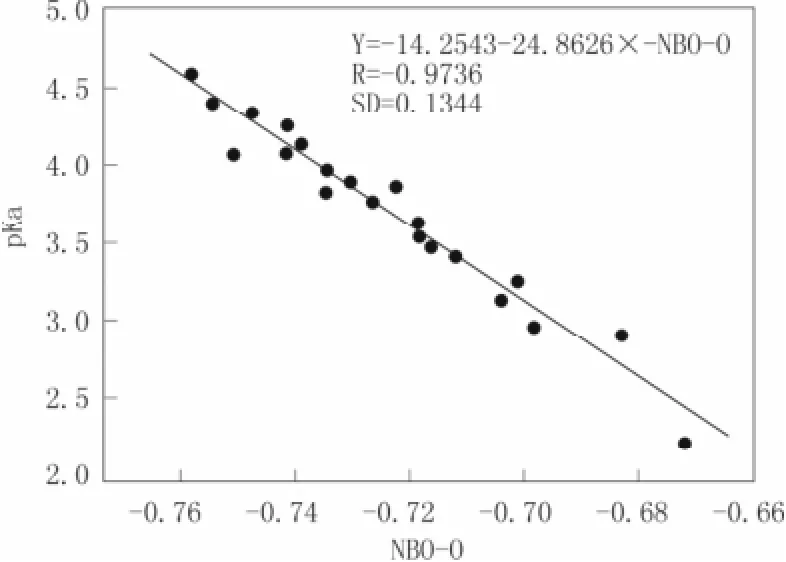
图 3 羧基环 O 原子的 NBO 电荷与实验 pKa之间的关系Fig.3 The relation of experiment pKawith the NBO charges of the O atom
电子 密 度ρ以 及 由 它 算 出 的 电 荷(NBO,ESP等),其大小值是能反映分子酸碱性的。当然由于采用方法的水平、基组的大小,ρ及电荷反映客观存在的精确水平是有差别的(见图 4)。

图 4 苯甲酸分子的原子电荷Fig.4 Atomic charges of benzoic acid
进一步的研究发现,该预测方法所使用的自然原子轨道电荷 (NBO) 值与所使用的泛函和基组以及溶剂关系不大。这预示着新方法可以使用分子的气相分子的优化数据来估测更加复杂衍生物分子的酸碱性。这为多取代苯甲酸药物类分子设计提供了便利。
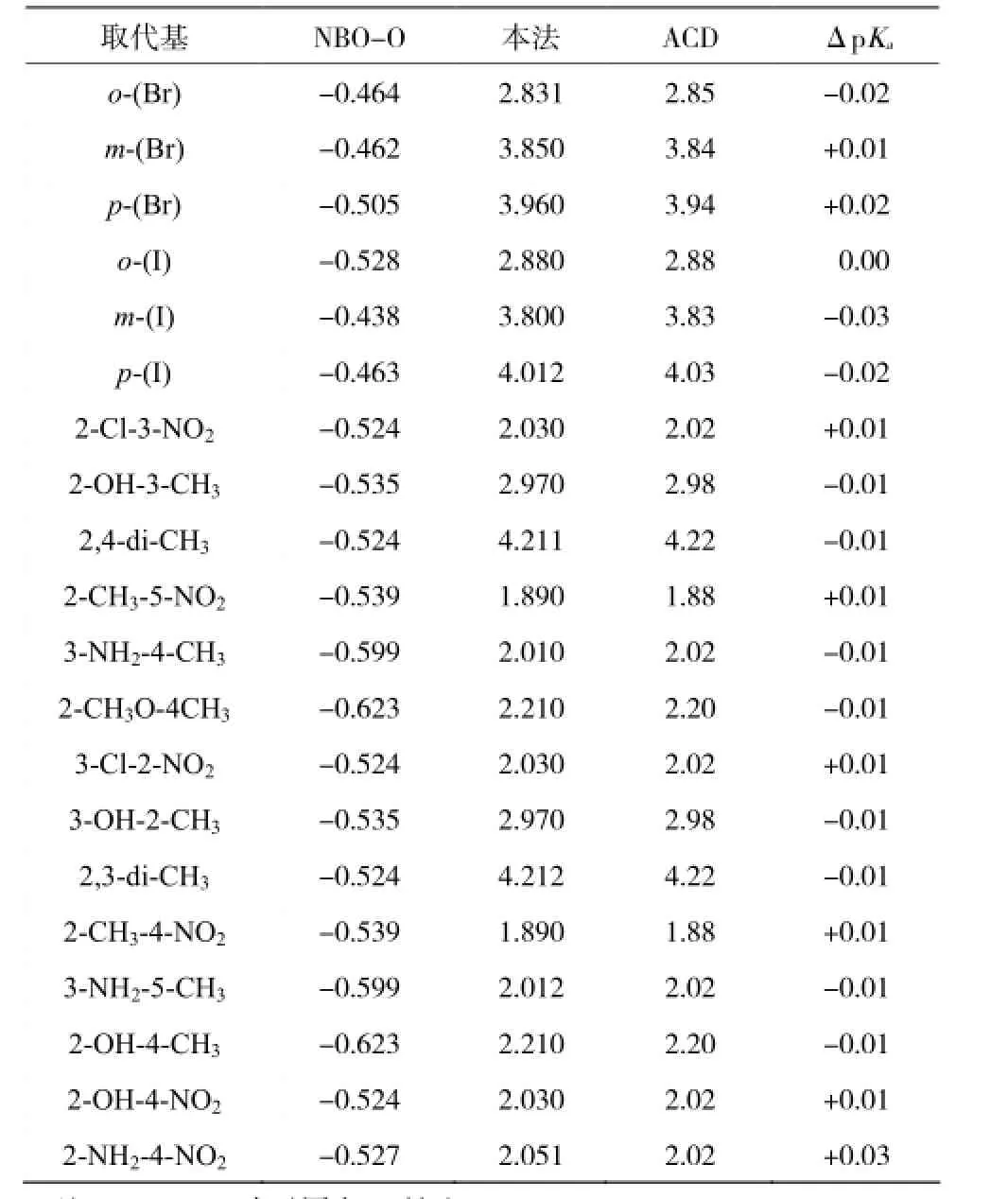
表 2 两种方法预测 2 0 种未知单及多取代苯甲酸 p Ka值Table 2 NBO parameter of 20benzoic acid with unknown pKa
3 结 论
(1)通过 Gaussion 09W 软件,使用密度泛方法 DFT/B3LYP/3-21+G(d),优化了 21 种单取代苯甲酸及其多取代苯甲酸分子结构,发现苯环羧基 O原子的 NBO-O 电荷值与其实验 pKa值的线性相关性较佳(Y=-14.254 3 - 24.862 6×NBO-O,相关系数R=-0.978 6)。
(2)计算了 12 个未知 pKa值的单和多元取代苯甲酸的 NBO 值,代入拟合出的较佳线性方程, 发现与 ACD-Labs 6.0 预测得到的 20 种取代苯甲酸分子 pKa实验值非常接近,最大偏差 ΔpKa< ±0.03。
[1]杨伟华,冯长君. 羧酸类化合物 pKa的定量构效关系[J]. 吉首大学学报(自然科学版), 2006,27(4):84-88.
[2]齐玉华,许禄.应用量化参数和 CoMFA 法研究苯甲酸类化合物的结构和其 pKa值的相关性[J].应用化学,2002,19(11):1489-1493.
[3]舒元梯. 取代酚酸性的分子拓扑研究[J].西南民族大学学报(自然科学版),2003(6):74-76.
[4]舒元梯. 取代苯甲酸酸性 pka的分子拓扑研究[J]. 达县师范高等专科学校学报(自然科学版), 2006,3(2):32-35.
[5]王超,冯长君.取代羧酸 pKa的分子树拓扑研究[J].化学研究,2004,15(2):67-69.
[6]刘良红, 张鹏飞, 黄莺. 用密度泛函活性理论和 Hammett常数预测单双取代苯酚的酸性[J]. 物理化学学报, 2013, 29 (3):508-515.
Estimating pKaValues of the Substituted Benzoic Acid by Natural Atomic Orbital Charge
LIU En-rong,ZHANG Yang-bin,ZHONG Ai-Guo
(College of Chemical Engineering and Pharmacy, Taizhou University, Zhejiang Taizhou 316000,China)
Density functional theory DFT B3LYP/3-21+G(d) basis set were used to optimize the molecular structure of 21 kinds of benzoic acid and substituted benzoic acid, it’s found that the natural atomic orbital charge value of O atom on the benzoic acid has good linear relativity with its experimental pKavalue, generally has better fitting result than its charge value of the electrostatic potential (ESP). NBO parameters of 20 substituted benzoic acid compounds with unknown pKavalues were calculated, they were substituted into the fitted linear parametric equation, it was found that the computed results were very close to substituted benzoic acid pKavalue predicted by the popular software ACD-Labs 6.0.
Density functional theory; Substituted benzoic acid; Natural atomic orbitals charge; PKa
O 621
: A
: 1671-0460(2014)03-0324-03
浙江省大学生科技创新项目(新苗人才计划),项目号:2014326。
2014-01-20
刘恩榕(1992-),男,浙江新昌人,现从事计算工作。
钟爱国(1964-),男,教授,硕士,研究方向:计算化学。E-mail:zhongaiguo@tzcedu.cn。
猜你喜欢
分子催化(2022年1期)2022-11-02
云南化工(2020年11期)2021-01-14
纺织科学与工程学报(2020年1期)2020-06-12
中成药(2018年12期)2018-12-29
中南大学学报(自然科学版)(2016年2期)2017-01-19
百科知识(2016年22期)2016-12-24
当代化工研究(2016年5期)2016-03-20
应用化工(2014年10期)2014-08-16
应用化工(2014年7期)2014-08-09
天然产物研究与开发(2014年7期)2014-04-27
