
西藏藏族结核分枝杆菌临床分离株的MIRU-VNTR基因多态性分析与评价
2014-04-08 01:11:30董海燕索朗德吉赵秀芹万康林
中国人兽共患病学报 2014年2期
石 荔,董海燕,巴 桑,西 洛,索朗德吉,赵秀芹,万康林
西藏是中国结核病疫情最严重的地区之一。本研究从来自西藏7个地区的藏族结核病患者中共分离出577株结核分枝杆菌,应用欧盟推荐的24位点MIRU-VNTR基因分型方法进行分子流行病学研究,并分析了24个VNTR位点的分辨率,旨在了解西藏藏族结核病患者中结核分枝杆菌的基因多态性、主要流行菌株以及现行的24位点MIRU-VNTR基因分型方法是否适合西藏藏族结核病患者的分子流行病学研究。
1 材料与方法
1.1菌株 自577例西藏藏族结核病患者分离出577株结核分枝杆菌临床分离株,其中拉萨305株,日喀则80株,那曲49株,昌都46株,山南45株,林芝44株,阿里8株。由中国疾病预防控制中心传染病预防控制所传代培养、保存。
1.2结核分枝杆菌标准菌株H37Rv以及牛分枝杆菌BCG购自中国药品生物制品检定所,由中国疾病预防控制中心传染病预防控制所传代培养、保存。
1.3提取细菌DNA(CTAB法)[1]。
1.4位点选择 从http://minisatellites.u-psud.fr/网站记载的结核分枝杆菌基因组多态性重复序列的位点引物序列,选择欧盟推荐的24个VNTR位点(ETR-A、ETR-B、ETR-C、ETR-D(MIRU04)、ETR-E(MIRU31)、MIRU02、MIRU10、MIRU16、MIRU20、MIRU23、MIRU24、MIRU26、MIRU27、MIRU39、MIRU40、Mtub04、Mtub21、Mtub29、Mtub30、Mtub34、Mtub39、Qub26、Qub11-b及Qub4156c)。
1.5PCR反应体系及扩增 总体积为15 μL。即2×Taq Master Mix 7.5 μL,引物( 5 μmol/L) 各1.5 μL,DNA模板 2.5 μL,去离子水 2.0 μL。PCR预变性95 ℃,15 min;变性94 ℃,1 min;退火60 ℃,1 min;延伸72 ℃,1.5 min;35个循环。最后延伸72 ℃,10 min。PCR产物4 ℃保存。
1.62%琼脂糖凝胶电泳
1.7位点重复次数计算 通过DNA Marker和标准菌株H37Rv的VNTR位点重复单元的重复次数作标准参照,根据扩增片段大小利用BioNumerics5.0软件计算出被测菌株各VNTR位点重复单元的重复次数。
1.8数据处理分析 实验数据及结果数字化后用Excel录入,登录网站http://www.miru-vntrplus.org比对分析。应用BioNumerics5.0软件对分型结果进行聚类分析。同时应用SPSS13.0软件对数据进行统计学分析。
1.9Hunter-Gaston指数的计算,公式如下:
其中,N为菌株总数。S为总类型数,nj是第j类型的菌株数。
2 结 果
2.124个位点MIRU-VNTR基因多态性分析 将577株结核分枝杆菌MIRU-VNTR基因分型结果转化成数字,用BioNumerics5.0软件进行聚类分析(图1),577株结核分枝杆菌呈现出347种基因型,其中278株表现为独特的基因型,另299株分为69个基因簇(2-37株菌),成簇率为51.82%。对于所有分析的菌株,24位点的MIRU-VNTR分型方法的Hunter-Gaston指数是0.992。
2.224个MIRU-VNTR位点等位基因多态性分析 本研究选择的24个VNTR位点中,Qub26位点有27株结核分枝杆菌没有扩增产物;Qub11b位点有28株结核分枝杆菌没有扩增产物;Qub4156c有21株结核杆菌没有扩增产物;MIRU16位点有7株结核分枝杆菌没有扩增产物;MIRU26位点有3株结核分枝杆菌没有扩增产物。计算各位点的Hunter-Gaston指数(表1),发现无论是对所有分析的菌株还是对北京家族菌株进行基因分型,MIRU31和Qub11b位点的分辨率很高(Hunter-Gaston指数≥0.6),Qub26、 Qub4156c、Mtub21、MIRU26和MIRU20位点的分辨率适中(Hunter-Gaston指数≥0.3),而其它位点的分辨率较差(Hunter-Gaston指数<0.3)[2],尤其是MIRU24位点的Hunter-Gaston指数为0。

表1 24个VNTR位点对所有菌株及其中北京家族菌株的Hunter-Gaston指数分辨率

图1577株结核分枝杆菌MLVA-24最小生成树图
每个圆形代表一种基因型,圆形的大小代表菌株数量的多少
Fig.1TheminimumspanningtreeofMLVA-24of577M.tuberculosis
Each circle represents a genotype and the size of the circular representative strains quantity.
2.3与数据库的比对分析 将577株结核分枝杆菌的24个位点MIRU-VNTR分型结果在http://www.miru-vntrplus.orgMIRU-VNTRplus数据库进行聚类分析,采用UPGAMA法,聚类分析结果显示,西藏藏族结核分枝杆菌主要包括4个基因家族的菌株,分别为北京家族菌株、T家族菌株、CAS家族菌株和LAM家族菌株(图2)。
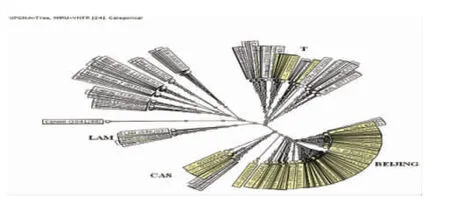
图2577株结核分枝杆菌MLVA结果与数据库菌株聚类结果,黄色方框代表本研究中的菌株,白色方框代表数据库中的菌株。
Fig.2ClusteringandMLVAresultsof577strainsofM.tuberculosistogetherwithdatabase.
The yellow boxes represent strains in this study and White Square on behalf of the strains in the database.
3 讨 论
采用欧盟推荐的24位点MIRU-VNTR分型方法对577株结核分枝杆菌进行了分型,结果显示577株结核分枝杆菌呈现出347种基因型。与MIRU-VNTRplus数据库中的菌株进行比对分析,发现577株结核分枝杆菌可聚成4个基因簇。其中最大的一簇为北京家族菌株,含523株,占90.64%(523/577)。这与曾报道过的西藏地区北京家族占90.28%相一致[3]。进一步表明了西藏藏族结核病患者中的流行优势菌是北京家族菌。而在欧洲,北京家族菌株的比例为16%,在美国的比例为4%[4], 因此欧盟推荐的方法是否适用于北京家族菌株流行区的分型研究尚待进一步探讨及评价。
欧盟之所以推荐使用24位点的MIRU-VNTR分型方法,是因为认为这些位点组合的分辨力与IS6110-RFLP相似。本研究中,其中7个VNTR位点(Qub11b、MIRU31、Qub26、 Qub4156c、Mtub21、MIRU26和MIRU20)对西藏藏族结核分枝杆菌的基因分型分辨率较高,其Hunter-Gaston指数均大于0.3,表明这7个位点的多态性较好,适用于对北京家族为主的结核分枝杆菌的基因分型。其它17个VNTR位点的基因多态性较差,其Hunter-Gaston指数小于0.3,表明这些位点不适用于对以北京家族为主的西藏藏族结核分枝杆菌进行基因分型。MIRU24位点相对保守,利用该位点可以区分古典型结核分枝杆菌(含有TbD1区)和现代型结核分枝杆菌(不含有TbD1区)[5],MIRU24位点的重复次数≥2的菌株为古典型结核分枝杆菌,重复次数为1的菌株为现代型结核分枝杆菌。本研究中MIRU24位点的重复次数均为1,提示西藏藏族结核病患者中没有发现古典型结核分枝杆菌,表明西藏是一个较现代的结核病的流行地区。MIRU26位点在北京家族菌株中的分辨指数为0.430,大于0.3,说明该位点适合北京家族菌株的分型研究。Rao KR等研究认为北京家族菌株在MIRU26位点具有7个重复片段,因此认为该位点可用于北京家族菌株的快速鉴定[6]。而本研究中北京家族菌株在MIRU26位点有出现7个重复片段,也有多于7个和少于7个重复片断的菌株,一些非北京家族菌株在该位点也具有7个重复片段。从而表明了同一VNTR位点不同地区的北京家族菌株的重复次数不一样,因此北京家族菌株的MIRU-VNTR分型具有多样性。
本研究西藏藏族结核病患者结核分枝杆菌基因多态性,并且北京家族菌株为主要的流行菌株,24位点MIRU-VNTR分型方法在对北京家族菌株进行分型研究。其中的一些位点的分型效果较差。这与日本的研究结果一致,并且日本也提出了适合北京家族菌株流行地区的12位点组合的VNTR分型方法[7]。由于不同地区的结核分枝杆菌的基因多态性及主要流行菌株并不相同,而且不同地区的北京家族菌株也具有不同的VNTR型。因此在对不同地区的结核分枝杆菌进行分子流行病学研究时,要考虑不同的VNTR位点组合,必要时要加入新的VNTR位点以增加基因分型的分辨力。
参考文献:
[1]Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual[M]. 2nded. New York: Cold Spring Harbor Laboratory Press, 1989.
[2]van Embden JD, Cave MD, Crawford JT, et al. Strain identification ofMycobacteriumtuberculosisby DNA fingerprinting: recommendations for a standardized methodology[J]. J Clin Microbiol, 1993, 31(2): 406-409.
[3]Shi L, Yang M, Pource C, et al.Genotyping research of 216 strains ofMycobacteriumtuberculosisclinical isolates in the Tibetan areas using MLVA and spoligotyping[J]. Chin J Microbiol Immunol, 2007, 27: 711-718. (in Chinese)
石荔,杨敏,Pource C, 等. MLVA和Spoligotyping 用于西藏地区216株结核分枝杆菌临床分离株的基因分型研究[J].中华微生物和免疫学杂志,2007,27:711-718.
[4]Filliol L, Driscoll JR, Van soolingen D, et al. Global distribution ofMycobacteriumtuberculosisspoligotypes[J]. Emerg Infect Dis, 2002, 8(11): 1347-1349. DOI: 10.3201/eid0811.020125
[5]Sun YJ, Bellamy R, Lee AS, et al. Use of mycobacterial interspersed repetitive unit-variable-number tandem repeat typing to examine genetic diversity ofMycobacteriumtuberculosisin Singapore[J]. J Clin Microbiol, 2004, 42(5): 1986-1993. DOI: 10.1128/JCM.42.5.1986-1993.2004
[6]Rao K, Ahmed N, Srinivas S, et al. Rapid identification ofMycobacteriumtuberculosisBeijing genotypes on the basis of the mycobacterial interspersed repetitive unit locus 26 signature[J]. J Clin Microbiol, 2006, 44(1): 274-277. DOI: 10.1128/JCM.44.1.274-277.2006
[7]Murase Y, Mitarai S, Sugawara L, et al. Promising loci of variable numbers of tandem repeats for typing Beijing familyMycobacteriumtuberculosis[J]. J Clin Microbiol, 2008, 57(7): 873-880. DOI: 10.1099/jmm.0.47564-0
猜你喜欢
西部医学(2024年3期)2024-03-21 12:22:24
保健医苑(2022年5期)2022-06-10 07:46:46
基层中医药(2020年5期)2020-09-11 06:32:00
基层中医药(2018年5期)2018-08-31 02:35:42
特别健康(2018年4期)2018-07-03 00:38:26
中国医学装备(2016年6期)2016-12-01 06:44:41
中国卫生(2015年1期)2015-11-16 01:06:02
制造技术与机床(2015年10期)2015-04-09 07:06:14
中国当代医药(2015年36期)2015-03-11 20:03:30
结核与肺部疾病杂志(2014年1期)2014-07-18 11:09:32
