
14-去甲基自溶霉素C11~C15片段的合成研究
2014-03-27 02:02刘许歌沈悦海
云南民族大学学报(自然科学版) 2014年3期
冯 亮,杨 帆,刘许歌,沈悦海
(昆明理工大学 生命科学与技术学院,云南 昆明 650500)
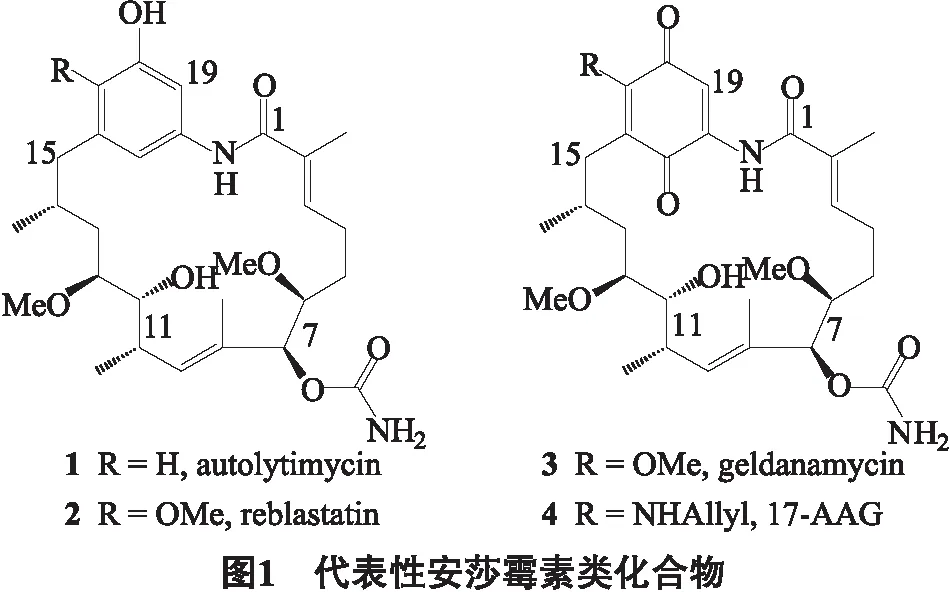
自溶霉素1(Autolytimycin)是2000年被发现于云南的一种聚酮类大环内酰胺天然产物[1].结构上属于安莎霉素(见图1),其类似物多具有较强的抗癌活性.例如格尔德霉素(3)及其处于临床试验阶段的半合成衍生物17-AAG(4).与结构3、4等苯醌型安莎霉素相比,以自溶霉素和Reblastatin(2)为代表的非醌型安莎霉素具有更好的安全性和类药性[2-3],因此其结构类似物的合成研究日益受到关注.Panek小组[4-5]分别报道了Reblastain和自溶霉素的全合成工作,Micalizio小组[6]报道了带有6-氨基甲酸酯基的自溶霉素类似物的合成和抗癌活性研究.最近,我们报道了自溶霉素C1~C8结构片段的合成研究[7].根据目前对安莎霉素构效关系的研究[8],自溶霉素分子结构的底部对于其结合靶蛋白Hsp90非常重要,而上半部分的贡献不大.因此,我们设想可除去自溶霉素的14-甲基,在保持化合物抗癌活性的同时,可以简化其结构和合成方法.
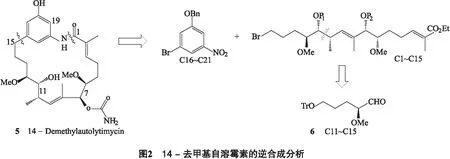
在14-去甲基自溶霉素(5)的逆合成分析中(图2),通过切断内酰胺和15,16-碳碳键,得到C16~C21苯环片段和C1~C15脂链片段,其中15,16-碳碳键可通过烷基芳基偶联反应构建.C1~C15片段可在10,11-碳碳键切断,以C11~C15片段(6)为合成起始砌块.在此,我们对C11~C15片段的合成研究进行报道.
1 实验部分
1.1 仪器和试剂
Buchi R-3旋转蒸发仪(瑞士步琪有限公司);IKA RCT basic 磁力搅拌器(广州仪科实验室技术有限公司);WZZ-2B自动旋光仪(上海申光仪器仪表有限公司);Bruker AV400核磁共振仪(德国布鲁克股份公司).
L-谷氨酸(上海新兴化工试剂研究所);硼烷二甲硫醚(Acros Organics);草酰氯、咪唑(中国阿拉丁试剂有限公司);亚硝酸钠、盐酸、氯仿(成都市科龙化工试剂厂);0.4 nm分子筛、无水硫酸镁、硼氢化钠、三氟化硼乙醚、吡啶、二甲亚砜、二氯甲烷、四氢呋喃、二甲基甲酰胺(国药集团化学试剂有限公司);叔丁基二甲基氯硅烷(北京偶合科技有限公司);三乙胺(天津市永大化学试剂有限公司);4-二甲胺基吡啶、四丁基氟化铵、Me3OBF4(上海阿达玛斯试剂有限公司);Proton-Sponge(Sigma-Aldrich有限公司);硅胶(青岛海洋化工厂分厂).
四氢呋喃经无水无氧处理;二甲亚砜、二氯甲烷和三乙胺经活化0.4 nm分子筛除水;石油醚(60~90 ℃)和乙酸乙酯经重蒸.
1.2 自溶霉素C11~C15片段的合成
14-去甲基自溶霉素C11~C15片段的合成路线见图3.
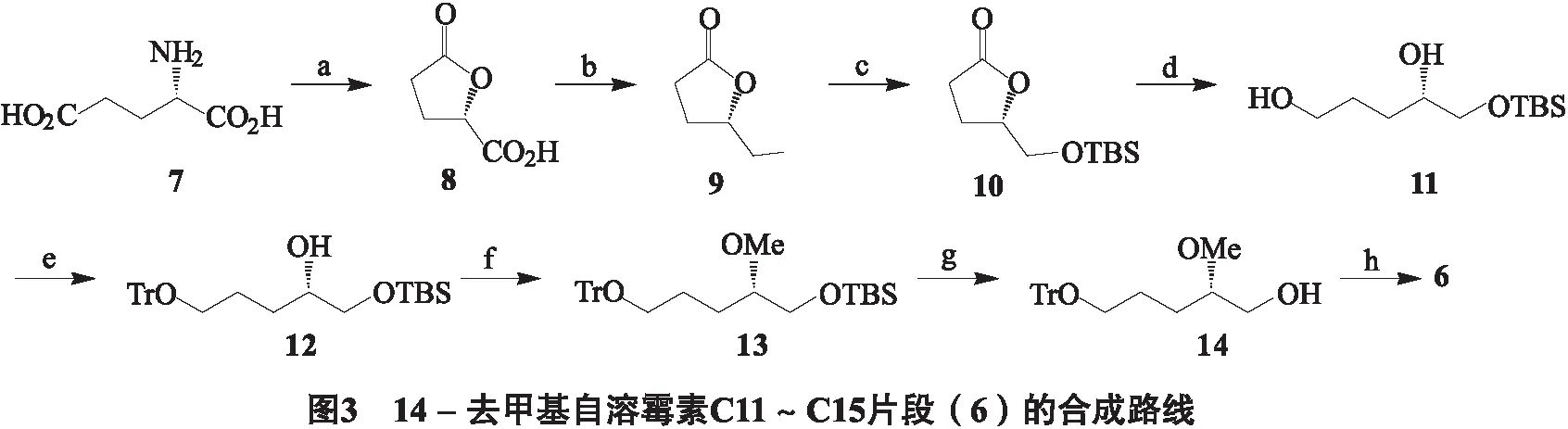
反应条件为a:NaNO2, HCl, 0~5 ℃; b:NaBH4, BF3·Et2O, 0 ℃; c:TBSCl, IMD, DMF, r.t.; d:BH3·Me2S, THF, 45℃; e:TrCl, Py, DMAP, DCM, r.t.; f:Proton Sponge, Me3OBF4, DCM, r.t.; g:TBAF, r.t.; h:oxalyl chloride, DMSO, Et3N, -78 ℃.
化合物9:取 1 000 mL三口圆底烧瓶,装入7(44.1 g, 0.3 mol)和水(120 mL),搅拌溶解,冷却到0~5 ℃后开始滴加盐酸(2 mol/L, 200 mL).滴加完毕后,维持温度在0~5 ℃,继续滴加NaNO2水溶液(0.42 mol, 29.3 g),控制在2 h滴完.随后将反应装置移至室温,搅拌12 h.继续补加盐酸(2 mol/L, 45 mL)和NaNO2水溶液(0.09 mol, 6.3 g),室温搅拌12 h.减压蒸去部分水,水相加入NaCl至饱和,EtOAc(800 mL)萃取,合并有机相,无水MgSO4干燥,过滤浓缩,得化合物8粗产物,浅黄色油状物27.2 g.
取烘干的 1 000 mL三口圆底烧瓶,装入NaBH4(0.32 mol, 12.4 g)和无水THF(220 mL),氮气保护,搅拌并降温至0 ℃,滴加上述油状物的无水THF溶液.待无气体冒出后,开始滴加BF3·Et2O(0.32 mol, 41 mL)的THF溶液(150 mL),控制在1 h左右滴完.滴加完毕后,反应混合物继续在0 ℃搅拌0.5~1 h,TLC监控反应.待反应完全,在0 ℃缓慢滴加水(15 mL)和甲醇(30 mL)淬灭反应,加完后升至室温,继续搅拌2~3 h,过滤,固体残余物用丙酮洗涤,合并液相和丙酮洗涤液,无水MgSO4干燥,过滤,减压浓缩,硅胶柱层析纯化,用V(PE)∶V(EA)=2∶1洗脱,得化合物9纯品,无色油状物9.7 g,2步总产率28%.
Rf=0.68,V(DCM)∶V(MeOH)=9∶1;1H NMR (400 MHz, CDCl3)δ:4.53~4.62 (m,1H), 3.85 (dd,J=12.5, 2.8 Hz, 1H), 3.59 (dd,J=12.5, 4.6 Hz,1H), 2.42~2.65 (m,2H), 2.03~2.26 (m, 2H).
化合物10:取100 mL单口圆底烧瓶,装入9(45 mmol,5.2 g)和DMF(25 mL),搅拌下冷却到0 ℃,加入咪唑(112.5 mmol,7.7 g)和TBSCl(58.5 mmol,8.8 g).加完后,升至室温搅拌3 h.加入饱和碳酸氢钠溶液(20 mL),用PE萃取(80 mL),萃取液用饱和食盐水洗涤,无水MgSO4干燥,过滤,硅胶柱层析纯化,用V(PE)∶V(EA)=9∶1洗脱,得化合物10纯品,无色油状物9.9 g,产率96%.
Rf= 0.57,V(PE)∶V(EA)=2∶1;1H NMR (400 MHz, CDCl3)δ:4.55~4.62 (m, 1H), 3.86 (dd,J=11.3, 3.2 Hz, 1H), 3.69 (dd,J=11.3, 3.1 Hz, 1H), 2.55~2.67 (ddd,J=17.5, 10.2, 7.3 Hz, 1H), 2.41~2.52 (ddd,J= 17.7, 10.0, 6.3 Hz, 1H), 2.12~2.32 (m, 2H), 0.89 (s, 9H), 0.07 (s, 6H).
化合物11:取250 mL三口圆底烧瓶,装入10(36 mmol, 8.3 g)和无水THF(50 mL),氮气保护下搅拌并冷却到0 ℃,滴加BH3·Me2S(54 mmol, 5.4 mL),随后继续在0 ℃搅拌30 min,再缓慢升温至45 ℃,搅拌过夜,TLC监控确定反应终点.缓慢滴加甲醇(50 mL)淬灭反应,减压旋蒸,硅胶柱层析纯化,用V(PE)∶V(EA)=5∶1~2∶1洗脱,得化合物11纯品,无色油状物8.3 g,产率98%.
化合物12:取100 mL单口圆底烧瓶,装入11(20 mmol, 4.7 g)和DCM(45 mL),室温搅拌,加入三苯基氯甲烷(22 mmol, 6.2 g),吡啶(40 mmol, 3.2 mL)和DMAP(0.4 mmol, 0.049 g).室温搅拌12 h后,加入饱和氯化铵溶液(20 mL)淬灭反应,盐酸(0.2 mol/L, 100 mL)洗涤,水相用EtOAc(200 mL)萃取,合并有机相,饱和食盐水洗,无水MgSO4干燥,过滤,浓缩,硅胶柱层析纯化,用V(PE)∶V(EA)=40∶1~30∶1,得化合物12纯品,无色油状物8.9 g,产率93%.
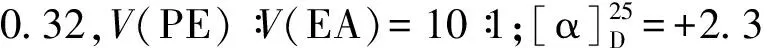
化合物13:取烘干的500 mL单口圆底烧瓶,装入12(11 mmol, 5.2 g)和DCM(150 mL),搅拌溶解,加入0.4 nm分子筛粉末(12.0 g),质子海绵(33 mmol, 7.1 g)和Me3OBF4(27.5 mmol, 4.2 g).室温搅拌18 h后,反应液经硅藻土过滤,盐酸(0.2 mol/L, 200 mL)洗涤,水相用EtOAc(80 mL)萃取,合并有机相,用饱和食盐水洗涤,无水MgSO4干燥,过滤,浓缩,硅胶柱层析纯化,用V(PE)∶V(EA)=40∶1,得无色油状的化合物13纯品(3.1 g)和原料12(1.8 g),扣除回收原料后产率为89%.
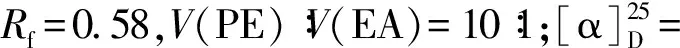
化合物14:取100 mL单口圆底烧瓶,装入13(4 mmol, 2.0 g)和THF(40 mL),搅拌溶解,0 ℃下缓慢加入TBAF(1 mol/L,8 mmol,8 mL),在0 ℃继续搅拌30 min,再升至室温搅拌5 h.减压蒸去溶剂,硅胶柱层析纯化,用V(PE)∶V(EA)=5∶1~3∶1,得化合物14纯品,无色油状物1.4 g,产率93%.
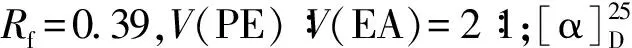
化合物6:取烘干的100 mL三口烧瓶,装入草酰氯(4 mmol,0.35 mL)和DCM(8 mL),降温至-78 ℃,加入DMSO(8 mmol,0.58 mL),在-78 ℃搅拌15 min,随后加入化合物8(0.38 g,1 mmol)的DCM溶液(4 mL),继续在-78 ℃搅拌20 min后加入TEA(15 mmol,2.1 mL),自然升温至10 ℃,加入水(15 mL),分出有机相,水相用DCM萃取(30 mL),合并有机相,MgSO4干燥,过滤,浓缩,硅胶柱层析纯化V(PE)∶V(EA)=10∶1~5∶1洗脱,得化合物6纯品,无色油状物0.36 g,产率91%.
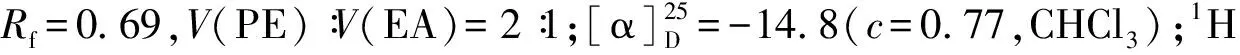
2 结果与讨论
对14-去甲基自溶霉素C11~C15片段的合成工作以L-谷氨酸(7)作为起始原料,经济高效地实现了14-去甲基自溶霉素C11~C15片断(6)的合成.值得注意的是,Panek小组[5]报道的自溶霉素C3~C7片段与这一片段结构类似,因此在硅醚10的合成中计划采用类似的方法,即在重氮化条件下将7转化为内酯酸8,随后经羧基还原和硅基保护得到10.通过对工艺的改进,以较高产率得到了化合物10.
在内酯酸8的制备中,发现后处理较为困难.8的水溶性较大,曾尝试用四氢呋喃进行萃取,虽然萃取迅速,但会在粗产物中带入大量氯化钠,不利于后续反应的计量;由于8极性很大,难以通过柱层析方法进行克级以上的纯化备料.最后我们发现用乙酸乙酯多次萃取效果较好,萃取液经干燥浓缩,得到的粗产物可直接用于下步反应,粗产率为70%.
在将内酯酸8还原为醇9时,Panek小组采用了较为昂贵的BH3·Me2S进行还原.由于内酯酸8粗产物中含有的杂质明显增加了BH3·Me2S用量,为能较为经济地大量制备醇9,尝试改用价廉的NaBH4和BF3·Et2O体系[9-10].对NaBH4和BF3·Et2O的用量、BF3·Et2O的滴加温度、滴加后的反应温度和反应时间等参数进行了详细的考察,最终发现在NaBH4和BF3·Et2O用量均为1.5 eq,滴加温度和反应温度为0 ℃可得到约40%的稳定产率.在BF3·Et2O滴加过程中,由于释放大量气体,需严格控制滴加速度和反应温度,以免物料溢出.反应完毕后,需加少量水淬灭过量的NaBH4,再加甲醇处理得到产物溶液.

参照对羧酸硼烷还原的相关研究[11],对上述NaBH4/BF3·Et2O还原反应的可能机理进行了分析(图3).在此体系中,羧酸8首先与NaBH4反应,生成酰氧基硼酸盐A,并伴随氢气放出.此后随着BF3·Et2O的加入,A首先与BF3·Et2O作用,形成中间体B,再与体系中原位生成的BH3反应,被还原成中间产物六元环硼酸酐C,最后经水或甲醇水解得到产物9.对内酯醇9进行硅基保护[5, 13],以96%的产率得到TBS醚10.在选择保护基时,我们曾试图用苄基保护,但在NaH/BnBr[12]等常用条件下得到的产率均较低(47%~53%).化合物10经BH3·Me2S还原[14]得到开环二醇11,后处理简便,产率达98%.使用TrCl成功实现了二醇11的选择性保护[13],产率为93%.在此反应中,加入催化量的DMAP可有效缩短反应时间.需要指出的是,由于过量的TrCl在后处理过程中生成与产物极性非常接近的TrOH,很难通过柱层析完全除去,但不影响后续反应.在此,使用体积较小的保护试剂则难以区分伯羟基和仲羟基,如在NaH/BnBr条件下得到的是无法分离的2种苄基化产物.
在随后的仲醇12的甲基化反应中,我们发现常用的MeI/NaH体系不适用,由于反应中生成的仲醇负离子与TBS醚邻近,引起TBS的迁移,即布鲁克重排[15],实际得到的是预期产物13和硅基迁移产物的混合物.经改进,采用了碱性较弱的质子海绵/Me3OBF4[4],尽管在此条件下反应很难进行完全,但可完全避免上述副反应,以89%的产率实现甲基醚13的有效制备.甲基醚13经四丁基氟化铵[16]处理脱去TBS保护基,再经Swern氧化[14]顺利得到目标产物手性醛6,2步产率分别为93%和91%.HCl/MeOH体系也可用于脱去TBS,但对于化合物13会引起Tr保护基的部分脱除.至此,实现了14-去甲基自溶霉素C11~C15片段的合成.

参考文献:
[1] LI M G, WU S H, ZHAO L X, et al. Isolation and structure elucidation of autolytimycin, a new compound produced by Streptomyces Autolyticus JX-47[J].Chinese Chemistry Letter, 2001, 12 (10):903-906.
[2] ZHANG M Q, GAISSER S, NUR-E-ALAM M, et al. Optimizing natural products by biosynthetic engineering: Discovery of nonquinone Hsp90 inhibitors[J].Journal of Medicinal Chemistry, 2008, 51 (18):5494-5497.
[3] XIE Q, WONDERGEM R, SHEN Y, et al. Benzoquinone ansamycin 17AAG binds to mitochondrial voltage-dependent anion channel and inhibits cell invasion[J].Proceedings of the National Academy of Sciences, 2011, 108 (10):4105-4110.
[4] WRONA I E, GABARDA A E, EVANO G, et al. Total synthesis of reblastatin[J]. Journal of the American Chemical Society, 2005, 127 (43):15026-15027.
[5] WRONA I E, GOZMAN A, TOLDONE T, et al. Synthesis of reblastatin, autolytimycin, and non-benzoquinone analogues: Potent inhibitors of heat shock protein 90[J]. The Journal of Organic Chemistry, 2010, 75 (9):2820-2835.
[6] JESO V, CHERRY L, MACKLIN T K, et al. Convergent synthesis and discovery of a natural product-inspired paralog-selective Hsp90 inhibitor[J].Organic Letters, 2011, 13 (19): 5108-5111.
[7] YANG F, FENG L, WANG N Z, et al. Practical synthesis of C1e8 fragment of autolytimycin via a chelation-controlled diastereoselective addition of diisopropenylzinc to α-methoxy aldehyde[J].Tetrahedron, 2013, 69 (45),9463-9468.
[8] STEBBINS C E, RUSSO A A, SCHNEIDER C, et al. Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent[J]. Cell, 1997, 89 (2): 239-250.
[9] WALKER S D, BORTHS C J, DIVIRGILIO E, et al. Development of a scalable synthesis of a GPR40 receptor agonist[J].Organic Process Research & Development, 2011, 15 (3):570-580.
[10] CHO B T, YOON N M. Convenient procedure for the reduction of carboxylic acids via acyloxyborohydrides[J].Bulletin of Korean Chemical Society, 1982, 3 (4):149-152.
[11] LOBBEN P C, LEUNG S S W, TUMMALA S. Integrated approach to the development and understanding of the borane reduction of a carboxylic acid[J].Organic Process Research & Development, 2004, 8 (6):1072-1075.
[12] ELWORTHY T R, BRILL E R, CHIOU S S, et al. Lactams as EP4 prostanoid receptor agonists. 3. Discovery of N-ethylbenzoic acid 2-pyrrolidinones as subtype selective agents[J]. Journal of Medicinal Chemistry, 2004, 47 (25):6124-6127.
[13] CHAKRABORTY T K, SAMANTA R, KUMAR P K. A radical mediated approach to the stereoselective formal total synthesis of (+)-Sch 642305[J].Tetrahedron, 2009, 65 (34):6925-6931.
[14] HANESSIAN S, COOKE N G, DEHOFF B, et al. The total synthesis of (+)-ionomycin[J]. Journal of the American Chemical Society, 1990, 112 (13):5276-5290.
[15] BROOK A G. Molecular rearrangements of organosilicon compounds[J]. Accounts of Chemical Research, 1974, 7 (3):77-84.
[16] REDDY C R, DHARMAPURI G, RAO N N. Synthesis of the macrocyclic core of iriomoteolide 3A[J].Organic Letters, 2009, 11 (24):5730-5733.
猜你喜欢
中国酿造(2022年9期)2022-09-28
吉林农业(2019年6期)2019-06-11
食品与发酵工业(2019年3期)2019-03-01
教育教学论坛(2018年38期)2018-09-25
中成药(2017年10期)2017-11-16
中国调味品(2017年10期)2017-10-18
益寿宝典(2017年1期)2017-09-03
中成药(2017年4期)2017-05-17
中小学实验与装备(2016年1期)2016-04-19
红领巾·探索(2014年8期)2014-10-10
