
螺环结构膨胀单体的合成及膨胀性能研究
2014-03-25 10:09宁志高王长松
沈阳化工大学学报 2014年1期
邹 琳, 洪 臻, 宁志高, 王长松, 梁 兵
(沈阳化工大学 材料科学与工程学院, 辽宁 沈阳 110142)
作为商品化的热固性树脂在固化和使用过程中,由于分子的交联以及温度变化等原因都会使树脂产生体积收缩,当收缩不均匀时,就会在材料内部产生内应力.树脂中的残余应力是一种潜在的破坏因素,可导致树脂的机械强度下降,使树脂在使用过程中出现过早断裂,性能变劣等缺点.另外,由于体积收缩,也使它们在许多方面不能满足人们的特殊需要.例如,在某些工业应用场合,人们希望聚合物聚合前后的体积收缩率为零,如无内应力复合材料、浇铸树脂、高光洁度涂料、印模材料等;而在另外一些应用场合,则希望其在聚合过程中体积有所膨胀,如精密铸造、预应力塑料、高强度黏合剂、密封材料、填隙材料、补牙材料等,然而树脂固化时产生的体积收缩显然满足不了这些要求.为解决上述问题,膨胀单体的研究相继展开,由于开环聚合反应具有在聚合过程中产生体积膨胀的特点,可减少固化后因体积收缩带来的不良影响.螺环化合物因其可在阳离子引发下发生开环聚合反应,开环后可产生体积膨胀作用,被用以解决树脂中的体积收缩问题[1-7].
目前国外对膨胀单体的研究多基于对螺环原碳酸酯聚合方式的探索,特别是对光引发的膨胀单体聚合反应的研究颇多,其中膨胀单体多用多元醇与原碳酸四乙酯反应制备螺环化合物,研究其改性树脂的紫外光固化反应机理[8-13].此外,膨胀聚合反应的应用领域已被扩宽,其中涉及补牙材料[14-16],阻燃材料[17-20]以及微生物材料领域[21-24].膨胀单体和膨胀聚合反应已用于制备高强度复合材料、高性能黏合剂、生物降解高分子材料和医用高分子材料,也用于通用高分子材料的改性及合成带有官能团的低聚物等.
但在上述研究中尚存在两个问题,首选是合成的膨胀单体与预改性聚合物相容性较差,单纯依靠物理机械共混易导致共混不均,进而降低改性材料的机械性能;其次在合成膨胀单体研究中尚没有标准的测量膨胀率的方法.为解决上述存在的问题,本实验选用3-甲硫基丁醛与二正丁基氧化锡合成膨胀单体3,9-二羟甲基-3,9-二(二甲基乙硫基)-1,5,7,11-四氧杂螺环[5,5]十一烷,该单体与环氧树脂的相容性有所提高,改性后环氧树脂的机械性能也有所提高.实验选用国际标准GB1463中的浮力法对预聚体改性环氧树脂体积膨胀率进行测定,减少测量体积膨胀率的误差,供相关实验参考.
1 实验部分
1.1 试剂及仪器
3-甲硫基丁醛(质量分数98 %),石家庄利达化学品有限公司;二正丁基氧化锡(质量分数98 %),日照力德士化工有限公司;1,2-二氯乙烷、二硫化碳、无水氯化钙为分析纯,国药集团化学试剂有限公司;三氟化硼乙胺(质量分数为98 %),上海玛耀化学技术公司;乙酸乙酯、氢氧化钠、正己烷、甲苯、无水硫酸钠、三氯甲烷为分析纯,天津市博迪化工有限公司;乙酸(质量分数36 %),天津市博迪化工有限公司;甲苯-2,4-二异氰酸脂(TDI)(AR),天津福晨化学试剂厂.
核磁共振光谱仪(AVANCE Ⅲ 500 MHz),德国布鲁克公司;傅里叶红外光谱仪(NEXUS-470),美国热电公司;电子比重计(MH-20A),台湾Matsuhaku有限公司.
1.2 实验原理
膨胀单体3,9-二羟甲基-3,9-二(二甲基乙硫基)-1,5,7,11-四氧杂螺环[5,5]十一烷合成路线:
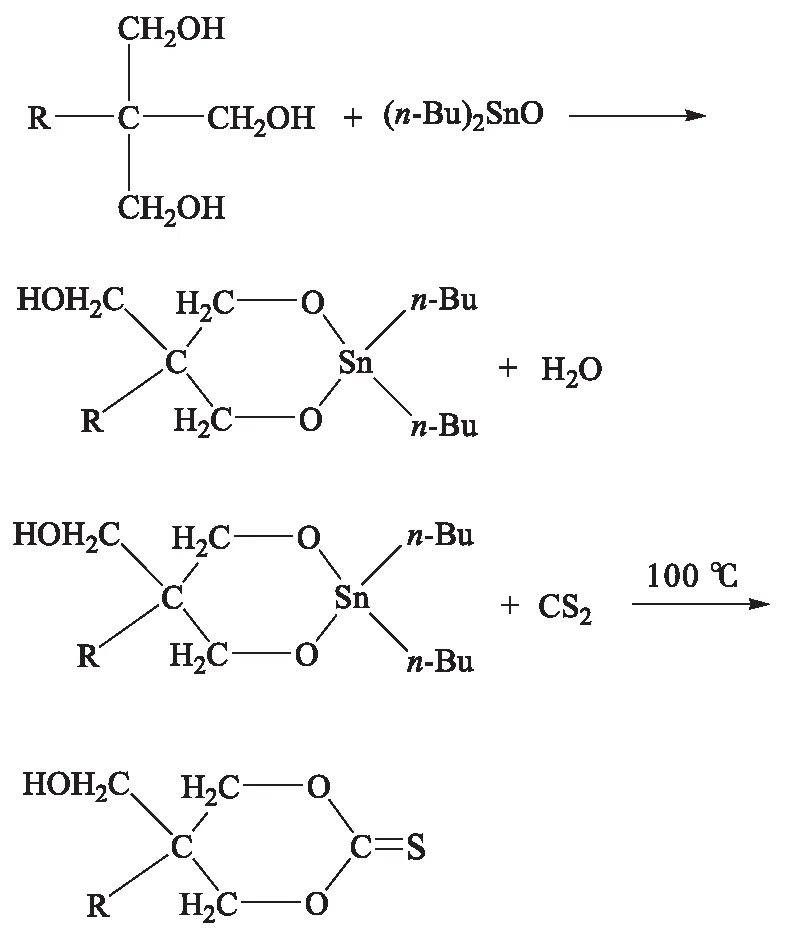
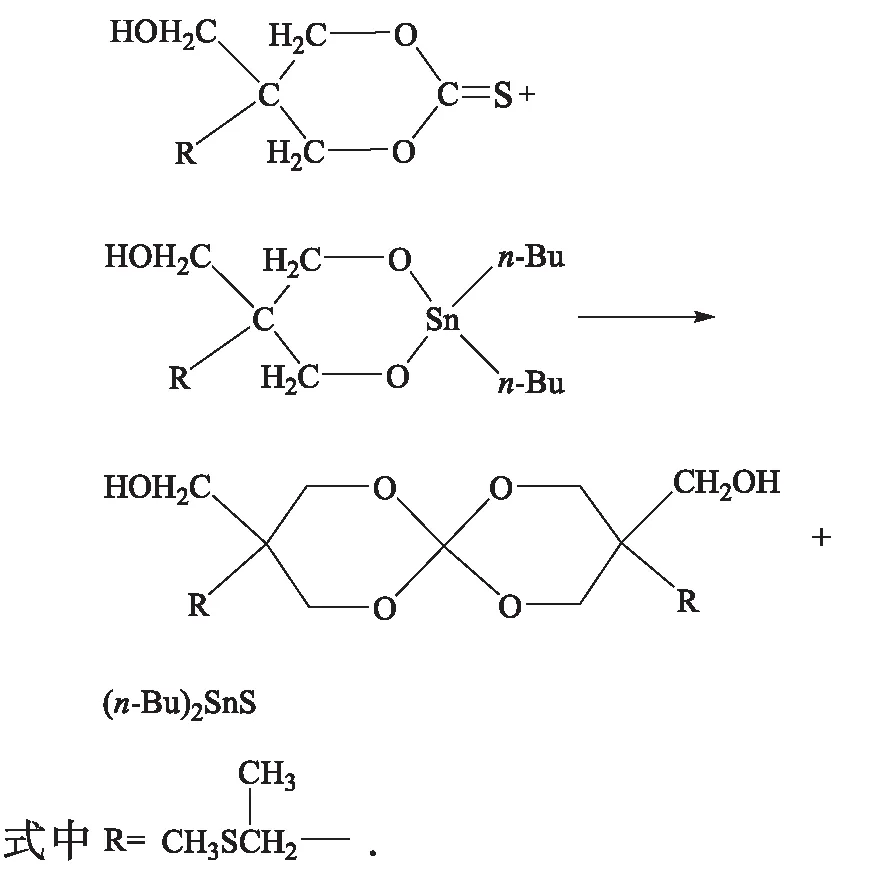
预聚体合成路线:
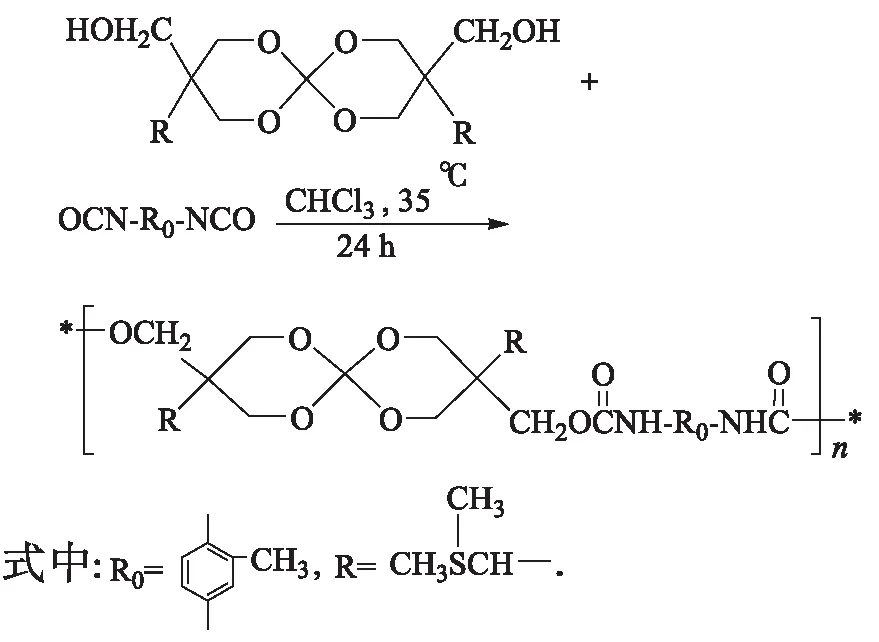
1.3 实验方法
1.3.1 三元醇的合成
在三口瓶中加入1 mol 3-甲硫基丁醛,4 mol甲醛水溶液和50 mL甲醇,电磁搅拌,缓慢滴加质量分数10 %的NaOH水溶液,控制pH值在10~11之间,反应2 h.反应完毕,滴加质量分数为36 %的乙酸溶液中和至pH=6,减压蒸馏除去大部分的水和过量的甲醛.用乙酸乙酯萃取,合并萃取液,用少量水洗涤,加无水硫酸钠干燥过夜.过滤除去硫酸钠,将滤液蒸馏以除去乙酸乙酯,再减压蒸馏即得产品,为浅黄色黏稠液体.
1.3.2 膨胀单体的合成
将0.1 mol三元醇,0.1 mol二正丁基氧化锡加入到三口瓶中,甲苯为溶剂,装上分水器和回流装置,电磁搅拌,130 ℃反应12 h.将油浴温度降至室温,拆除分水器,装上恒压滴液漏斗和回流装置,并在回流冷凝管上安装CaCl2干燥管.滴加8 mL CS2后升温至100 ℃,反应12 h.降至室温后,减压蒸馏出甲苯,用正己烷分数次洗涤以除去(n-Bu)2SnS,洗涤过程中继续搅拌,底部黏稠液体为较纯正的单体.用甲苯使其重结晶,干燥后制得膨胀单体,为褐色黏稠液体.
1.3.3 阳离子引发开环聚合体积膨胀率的测定
用膨胀计跟踪测量聚合过程中体系体积的变化.将质量分数3 %的三氟化硼乙胺催化剂加入到液态螺环单体中.真空烘箱中恒温60 ℃抽气至透明无气泡,将此试样放入膨胀计中,在恒温120 ℃的油浴中进行聚合反应,每10 min记录毛细管液面高度变化,直至高度不再变化时停止反应,冷却至室温读取毛细管内液面高度变化.根据膨胀率计算公式求得不同时间下的体积膨胀率,开环聚合体积膨胀率随时间变化趋势如图1所示.冷却后体积膨胀率为3.16 %.

图1 体积膨胀率随时间的变化曲线
1.3.4 预聚体的合成
分别将膨胀单体中羟基与TDI中氰酸基进行滴定,按反应官能团摩尔比为1∶1称量膨胀单体,加入氯仿将其溶解,另将TDI用10 mL氯仿溶解缓慢滴加到三口瓶中,35 ℃反应24 h.将产物倒入表面皿中,室温放置48 h,使溶剂自然挥发.
1.3.5 预聚体改性环氧树脂体积膨胀率的测定
实验中改性环氧树脂的体积收缩率采用密度法测定,此方法是分别测定固化前后树脂浇铸体密度,固化前树脂密度用比重瓶法测得,固化后树脂的密度测定可参照国标CB 1463中的浮力法进行测量.实验用电子比重计MH-20A分别测量树脂在空气中和在水中的质量.树脂浇铸体总体积收缩率计算根据公式:
计算求得.式中:Vs为树脂浇铸体总体积收缩率, %;ρo为固化前树脂系统密度,g/cm3;ρc为固化后树脂浇注体密度,g/cm3.
2 结果与讨论
2.1 三元醇的结构表征
采用溴化钾压片法进行测试.三元醇的红外光谱图如图2所示.1 028 cm-1处为伯醇的特征吸收峰,3 410 cm-1出现羟基吸收宽峰,与三元醇的结构相符.

图2 三元醇的红外光谱
核磁共振氢谱测试以DMSO为溶剂.三元醇的核磁共振氢谱如图3所示.1.38~1.50是甲基上的质子峰;2.18是与S相连的甲基上的3个H;3.0~3.25是与S相连的亚甲基上的质子峰;3.30~3.48是—CH2OH上的亚甲基上的6个H;4.78是—CH2OH上羟基的质子峰.

图3 三元醇的1H-NMR谱图
2.2 膨胀单体的表征
膨胀单体的红外谱图如图4所示.1 228 cm-1处为螺环特征吸收峰;1 729 cm-1处为碳酸酯羰基特征吸收峰;3 445 cm-1处为羟基特征吸收峰;2 900 cm-1左右为C—H伸缩振动吸收峰;1 457 cm-1和1 364 cm-1为伯醇的缔合特征吸收峰;1 016 cm-1为伯醇的C—O伸缩振动.

图4 膨胀单体的红外光谱
膨胀单体的1H-NMR谱如图5所示.1.23~1.42处为甲基上质子峰;2.00~2.10处为与S相连的甲基上的6个H;2.42~2.60处为与S相连的亚甲基上质子峰;3.20~3.51处为螺环上亚甲基上质子峰;4.08和4.30处为—CH2OH上的亚甲基的质子峰;4.78是—CH2OH上羟基的质子峰.
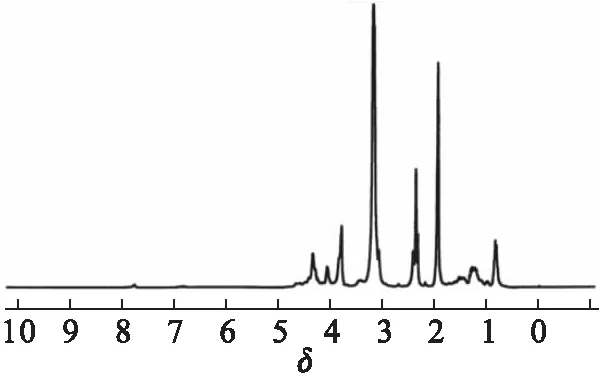
图5 膨胀单体的1H-NMR谱图
2.3 预聚体的表征
预聚体与膨胀单体的红外光谱如图6所示.从图6可以看到:预聚体在1 223 cm-1处螺环的特征峰仍然存在;1 732 cm-1处为碳酸酯羰基吸收峰;在3 334 cm-1附近为氨基特征吸收峰; 1 536 cm-1和1 599 cm-1为N—H弯曲振动;1 457 cm-1和1 364 cm-1处的伯醇缔合特征吸收峰明显减小.

图6 预聚体与膨胀单体的红外光谱图
2.4 预聚体改性环氧树脂体积膨胀率的测定
经实验测得改性环氧树脂的密度,通过公式计算出改性环氧树脂的体积变化率和预聚体体积膨胀率,如表1所示(膨胀为+,收缩为-).由表1数据可以看出:纯环氧树脂固化后出现体积收缩现象,体积收缩率为1.582 %,在相同固化条件下,随着体系中预聚体含量的增加,改性环氧树脂的体积膨胀率呈增大趋势.当预聚体质量分数为20 %时,改性环氧树脂的体积膨胀率为1.194 %.对于数值波动的原因分析可能为:膨胀单体预聚体膨胀率计算公式的前提是假设共聚树脂的体积变化是各个聚合物在相同条件下体积变化的线性加和,然而实际树脂的固化反应是一个非常复杂的交联过程,体系体积的变化可能还会受到其他因素的影响,因此对于预聚体膨胀率的计算会存在一些误差.

表1 改性环氧树脂的体积膨胀率
3 结 论
实验选用3-甲硫基丁醛与甲醛反应制得三元醇,用合成出的三元醇与二正丁基氧化锡反应制得螺环结构的膨胀单体3,9-二羟甲基-3,9-二(二甲基乙硫基)-1,5,7,11-四氧杂螺环[5,5]十一烷,产物经红外、核磁确定其分子结构.阳离子引发膨胀单体开环聚合体积膨胀率为3.16 %.预聚体改性环氧树脂体积膨胀率随体系中预聚体含量的增加,改性环氧树脂的体积膨胀率呈增大趋势,当预聚体质量分数为20 %时,改性环氧树脂的体积膨胀率为1.194 %.
参考文献:
[1] 袁金颖,潘才元.树脂固化时体积收缩内应力的本质及消除途径[J].化学与粘合,1998(4):234-240.
[2] HE P S,ZHOU Z Q,PAN C Y.An Epoxy Resin Copolymer with Zero Shrinkage[J].J Mater Sci,1989,24(5):1528-1532.
[3] 陈小庆,王岚,黄培,等.一种螺环原碳酸酯单体的合成及其与环氧树脂热固化[J].南京工业大学学报,2005,27(2):9-12.
[4] 于智,智放,王长松,等.一种新膨胀单体的合成及其预聚物与环氧树脂的共聚研究[J].沈阳化工大学学报,2003,17 (2):126-128.
[5] 王长松,周本濂.螺环原碳酸酯的预聚物与环氧树脂的共聚研究[J].高分子材料科学与工程,2000,16(1):28-31.
[6] 余红伟,王源升.膨胀单体研究进展[J].化学推进剂与高分子材料,2004,5(2):14-17.
[7] 王长松,潘才元.螺环原碳酸酯的预聚物对环氧树脂的改性研究[J].沈阳化工大学学报,1996,10(2):87-91.

[9] CHAPPELOW C C,PINZINO C S,CHEN S S,et al.Photopolymerization of a Novel Tetraoxaspiroundecane and Silicon-containing Oxiranes[J].Journal of Applied Polymer Science,2007,103(1):336-344.
[10] SANGERMANO M,GIANNELLI M,ORTIZ R A et al.Synthesis of an Oxetane-functionalized Hemispiroorthocarbonate Used as a Low-shrinkage Additive in the Cationic Ultraviolet Curing of Oxetane Monomers[J].Journal of Applied Polymer Science,2009,112(3):1780-1787.
[11] KUME M,MAKI Y,OCHIAI B,et al.Synthesis of Copolymers Containing a Spiro Orthocarbonate Moiety and Evaluation of the Volume Change during Their Cationic Crosslinking[J].Journal of Polymer Science:Part A:Polymer Chemistry,2006,44(24):7040-7053.
[12] STANSBURY J W,BAILEY W J.Synthesis and Evaluation of Spiro Orthocarbonate Monomers Capable of Polymerization with Expansion as Ingredients in Dental Composite Materials[J].Polymeric Materials Science and Engineering,1988,59:402-406.
[13] CHAPPELOW C C,PINZINO C S,CHEN S S,et al.Tetraoxaspiroalkanes for Polymerization Stress Reduction of Silorane Resins[J].Journal of Applied Polymer Science,2008,108(6):3738-3747.
[14] Yoo S H,Kim C K.Synthesis of a Novel Spiro Orthocarbonate Containing Bisphenol-A Unit and Its Application to the Dental Composites[J].MacromoLecular Research,2010,18(10):1013-1020.
[15] SUN X,LI Y C,XIONG J,et al.Shrinkage Properties of a Modified Dental Resin Composites Containing a Novel Spiro-orthocarbonate Expanding Monomer[J].Materials Letters,2011,65(23/24):3586-3589.
[16] WANG D Y,CAI X X,QU M H,et al.Preparation and Flammability of a Novel Intumescent Flame-retardant Poly(ethylene-Co-vinyl Acetate) System[J].Polymer Degradation And Stability,2008,93(12):2186-2192.
[17] VIJAYAKUMAR C T,MATHAN N D,SARASVATHY V,et al.Synthesis and Thermal Properties of Spiro Phosphorus Compounds[J].Journal of Thermal Analysis and Calorimetry,2010,101(1):281-287.
[18] CHEN D Q,WANG Y Z,HU X P,et al.Flame-retardant and Anti-dripping Effects of a Novel Char-forming Flame Retardant for the Treatment of Poly(ethylene terephthalate) Fabrics[J].Polymer Degradation and Stability,2005,88 (2):349-356.
[19] LI L K,WEI P,LI J,et al.Synthesis and Characterization of a Novel Flame Retardant and Its Application in Polycarbonate[J].Journal of Fire Sciences,2010,28(6):523-538.
[20] LIU Y,WANG D Y,WANG J S,et al.A Novel Intumescent Flame-retardant LDPE System and Its Thermo-oxidative Degradation and Flame-retardant Mechanisms[J].Polymers for Advanced Technologies,2008,19(11):1566-1575.
[21] SHARMA P,KUMAR A,SAHU V.Synthesis of Bioactive Spiro-2-[30-(20-phenyl)-3 hindolyl]-1-aryl-3-phenylaziridines and SAR Studies on Their Antimicrobial Behavior[J].Medicinal Chemistry Research,2009,18(5):383-395.
[22] AHADI S,ABASZADEH M,BAZGIR A.Organocatalytic Three-component Cascade Reaction for the Synthesis of Spiro[indeno[1,2-B]furan]-triones[J].Molecular Diversity,2012,16(2):299-306.
[23] KOUZNETSOV V V.Quinolines Spiro Annulated at Heterocyclic Fragment:Synthesis and Properties[J].Journal of Heterocyclic Chemistry,2005,42(1):39-59.
[24] KATO S,YOSHINO T,SHIBASAKI M et al.Catalytic Asymmetric Synthesis of Spirooxindoles by a Mannich-type Reaction of Isothiocyanato Oxindoles[J].Angewandte Chemie International Edition,2012,51(28):7007-7010.
猜你喜欢
应用化工(2022年4期)2022-06-22
中国酿造(2022年1期)2022-02-07
建筑材料学报(2018年1期)2018-03-07
苏州科技大学学报(自然科学版)(2017年1期)2017-03-20
中国塑料(2016年3期)2016-06-15
浙江大学学报(工学版)(2016年9期)2016-06-05
当代化工研究(2016年9期)2016-03-20
材料科学与工程学报(2016年5期)2016-02-27
中国塑料(2015年7期)2015-10-14
河南化工(2014年1期)2014-08-30
