
Critical illness polyneuropathy and myopathy: a systematic review
2014-03-23 07:19:03ChunkuiZhouLiminWuFengmingNiWeiJiJiangWuHongliangZhang
中国神经再生研究(英文版) 2014年1期
Chunkui Zhou, Limin Wu, Fengming Ni, Wei Ji, Jiang Wu Hongliang Zhang
1 Department of Neurology, the First Bethune Hospital, Jilin University, Changchun 130021, Jilin Province, China
2 Department of Neurology, the Second Part, the First Bethune Hospital, Jilin University, Changchun 130021, Jilin Province, China
3 Neuroprotection Research Laboratory, Massachusetts General Hospital, Harvard Medical School, Charlestown 02129, MA, USA
4 Department of Radiotherapy, Oncology Center, the First Bethune Hospital, Jilin University, Changchun 130021, Jilin Province, China
5 Department of Vascular Surgery, People’s Hospital of Jilin Province, Changchun 130000, Jilin Province, China
Critical illness polyneuropathy and myopathy: a systematic review
Chunkui Zhou1,2, Limin Wu1,3, Fengming Ni4, Wei Ji5, Jiang Wu1, Hongliang Zhang1
1 Department of Neurology, the First Bethune Hospital, Jilin University, Changchun 130021, Jilin Province, China
2 Department of Neurology, the Second Part, the First Bethune Hospital, Jilin University, Changchun 130021, Jilin Province, China
3 Neuroprotection Research Laboratory, Massachusetts General Hospital, Harvard Medical School, Charlestown 02129, MA, USA
4 Department of Radiotherapy, Oncology Center, the First Bethune Hospital, Jilin University, Changchun 130021, Jilin Province, China
5 Department of Vascular Surgery, People’s Hospital of Jilin Province, Changchun 130000, Jilin Province, China
Chunkui Zhou and Limin Wu contributed equally to this work.
Critical illness polyneuropathy and critical illness myopathy are frequent complications of severe illness that involve sensorimotor axons and skeletal muscles, respectively. Clinically, they manifest as limb and respiratory muscle weakness. Critical illness polyneuropathy/myopathy in isolation or combination increases intensive care unit morbidity via the inability or dif fi culty in weaning these patients off mechanical ventilation. Many patients continue to suffer from decreased exercise capacity and compromised quality of life for months to years after the acute event. Substantial progress has been made lately in the understanding of the pathophysiology of critical illness polyneuropathy and myopathy. Clinical and ancillary test results should be carefully interpreted to differentiate critical illness polyneuropathy/myopathy from similar weaknesses in this patient population. The present review is aimed at providing the latest knowledge concerning the pathophysiology of critical illness polyneuropathy/myopathy along with relevant clinical, diagnostic, differentiating, and treatment information for this debilitating neurological disease.
nerve regeneration; neurodegenerative diseases; critical illness polyneuropathy; critical illness myopathy; intensive care unit; sepsis; multiple organ failure; Guillain-Barré syndrome; NSFC grant; neural regeneration
Funding: This work was supported by grants from China Scholarship Council, No. 2008102056; the National Natural Science Foundation of China, No. 81241147.
Zhou CK, Wu LM, Ni FM, Ji W, Wu J, Zhang HL. Critical illness polyneuropathy and myopathy: a systematic review. Neural Regen Res. 2014;9(1):101-110.
Introduction
Critical illness polyneuropathy and critical illness myopathy are frequent complications of severe illness involving both motor and sensory axons. Critical illness polyneuropathy and myopathy usually present as flaccid and symmetric paralysis[1-2], which is seen in approximately 25–45% of critically ill patients who are admitted to intensive care units[3]. Limb (mostly from the lower extremities) and respiratory muscle weakness manifested by patients with critical illness polyneuropathy and myopathy often causes prolonged intensive care unit stays and mechanical ventilator dependence, in addition to increased long-term disability[4]. Weaning off mechanical ventilators is associated with problems attributed to involvement of the phrenic nerves, diaphragm, and intercostal and other accessory respiratory muscles[5]. Although rare, facial muscles can also be involved[6], ophthalmoplegia may occur[5]. Results from a multicenter Italian critical illness myopathy and/or neuropathy study showed that critical illness myopathy had a better prognosis compared with critical illness polyneuropathy[7]. Many patients continue to suffer from decreased exercise capacity and compromised quality of life for months to years after the acute event[7]. Recently, substantial progress has been made in the understanding of the pathophysiology of critical illness polyneuropathy and myopathy[1-3]. Both antiseptic prophylaxis and control of hyperglycemia may be effective for the prevention of critical illness polyneuropathy and myopathy; aggressive treatment of sepsis and multiple organ failure are considered to be the most effective measures to reduce the incidence[1-3].
Critical illness polyneuropathy and myopathy: history and risk factors
Neuromuscular dysfunction in the intensive care unit setting has attracted attention for over a century as it leads to high morbidity and poor prognosis[8]. Limb and respiratory muscle weakness in critically ill patients was initially attributed to catabolic myopathy and diaphragmatic fatigue, when it was fi rstly described in 1892 by Osler[8]. Later, in 1961, Mertens[9]demonstrated coma-related disseminated neuropathy and suggested metabolic and ischemic lesions of peripheral nerves as basic pathophysiological mechanisms. The term“critical illness polyneuropathy” was introduced in 1984 by Bolton et al.[10]who attributed characteristic axonal loss ofmotor and sensory fi bers to the toxic effect of sepsis. Critical illness myopathy, which has been increasingly recognized from the 1990s, can be elicited from excessive dosages of intravenous corticosteroids[11]. Increasing evidence in intensive care units has shown that critical illness polyneuropathy and myopathy frequently occur concomitantly[12].
Critical illness polyneuropathy affects more than one third of the most severe critically ill patients[13-14]; the percentage increases when associated with acute respiratory distress syndrome[15], sepsis, or systemic in fl ammatory response syndrome[16]and reaches up to 100% when further complicated by multiple organ failure[17]. Critical illness myopathy is present in 36% of those who need mechanical ventilation for severe asthma[18], and approximately 70% of patients who are admitted to intensive care units for at least 7 days, while most patients have critical illness polyneuropathy concomitantly[19]. However, the epidemiology tends to correlate with the duration and the severity of basic diseases[5,15,20-22].
As mentioned above, acute respiratory distress syndrome, sepsis, systemic in fl ammatory response syndrome, and multiple organ failure are the most common factors associated with either critical illness polyneuropathy or myopathy or a combination of both[14,23-24]. Prolonged bed rest, medication, and infections, are major risk factors for critical illness polyneuropathy and myopathy. In particular, gram-negative bacteremia is an independent risk factor for the development of critical illness polyneuropathy or myopathy. Hyperglycemia has also been identi fi ed as an independent risk factor[25], with important potential effects in terms of prevention[26]. Other less consistent risk factors include hyperpyrexia, hyperosmolality, hypoalbuminemia, hypoxia, hypotension, hyper/ hypocalcemia, female sex, old age, severe illness, central neurologic failure such as septic encephalopathy, long duration of organ dysfunction, renal failure and renal replacement therapy, parenteral nutrition, vasopressor and catecholamine support, and septic encephalopathy[16,24,27-31].
Pathophysiology of critical illness polyneuropathy and myopathy
Animal models have shown evidence of channelopathies in both critical illness polyneuropathy and myopathy, and human studies also identi fi ed axonal degeneration in critical illness polyneuropathy and myosin loss in critical illness myopathy[34-35]. The causes of axonal degeneration of sensory and motor axons in critical illness polyneuropathy, and atrophy and necrosis of myo fi bers in critical illness myopathy are complex and not fully understood; they possibly involve microcirculatory abnormality, metabolic derangements, reversible channelopathy, and bioenergetic dysfunction[36](Figure 1). Microvascular alterations including increased expression of E-selectin, vasodilatation, increased permeability of capillaries, extravasation, endoneural edema and hypoxemia; metabolic alterations including production of cytokines and other toxic factors, hyperglycemia, hormone imbalance, hypoalbuminemia, amino acid deficiency and activation of proteolytic pathways; electrical alterations including ion channel dysfunction, cell depolarization, inexcitability, altered Ca2+homeostasis and changes in excitation-contraction coupling; and bioenergetic failure including anti-oxidant depletion, reactive oxygen species increase, mitochondrial dysfunction, and apoptosis, all contribute independently, simultaneously, or synergistically to a combination of ischemic and cytopathic hypoxia and energy depletion, which eventually leads to critical illness polyneuropathy and myopathy[13,24]. Sepsis-related disturbances of the microcirculation in peripheral nerves and muscles, and the resultant production of cytokines, may increase the permeability of the microvasculature and further exacerbate hypoxemia and energy depletion[37]. Hyperglycemia[25], decreased ratio of anabolic/catabolic hormones[38], lower total amino acid levels[39], and hypoalbuminemia[5]can further increase endoneurial edema, which contributes to muscle weakness. Proteolytic pathways including calpain[40], lysosomal pathways, the ubiquitin-proteasome pathway[41-42], and transforming growth factor-β/mitogen-activated protein kinase pathways are up-regulated by in fl ammation and stress stimuli, inducing the breakdown of proteins (mainly by myofibrillar proteins such as actin and myosin), which are processed by the proteasome pathway[43-46]. Electrodiagnostic studies showed that sodium channels[47-51]and sarcoplasmic reticulum calcium release[35]play a vital role in muscle membrane hypoexcitability or inexcitability. The peripheral nerves of patients with critical illness polyneuropathy are depolarized, and membrane depolarization is related to endoneurial hyperkalemia or hypoxia, or both[52-53].
Acquired channelopathy involving voltage-dependent sodium channels and decreased expression of nitric oxide synthases results in hypoexcitability or inexcitability of muscle cell membranes[52]. Nitric oxide over-production is also associated with prolonged respiratory failure as it increases reactive oxygen species. Additionally, the immune system affects critical illness polyneuropathy and myopathy as proinflammatory and anti-inflammatory cytokines are released from in fi ltrating leukocytes in skeletal muscle[40-42]. Moreover, a thus far unknown low molecular weight neurotoxic agent has been identi fi ed in the serum of critical illness polyneuropathy patients. Specifically, an in vitro toxicity assay showed serum neurotoxicity in 12 of 16 patients with critical illness polyneuropathy[54]. A decreased ratio of anabolic/catabolic hormones contributes to myofilament loss and apoptosis[13]. Endogenous corticosteroid concentrations are temporarily increased at the beginning, but drop during the later phase because of adrenal insuf fi ciency[55].
Evaluation and diagnostic criteria for critical illness polyneuropathy and myopathy
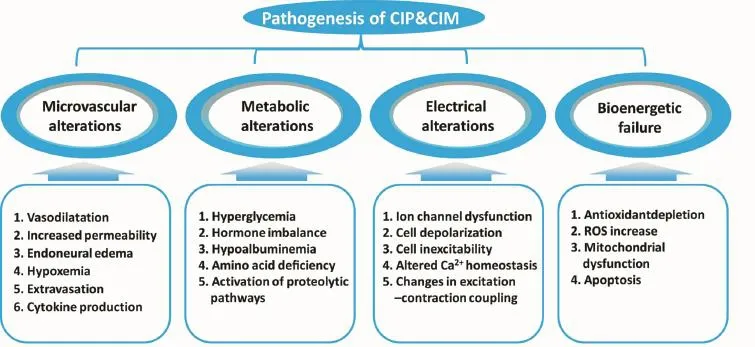
Figure 1 Pathogenesis of critical illness polyneuropathy (CIP) and myopathy (CIM).
The Medical Research Council sum score can be used as an initial diagnostic measure of muscle force in conscious patients who are suspected of having critical illness polyneuropathy or myopathy. Critical illness polyneuropathy and myopathy are arbitrarily diagnosed if the Medical Research Council sum score is less than 48[56]. Further investigations, including serum creatine kinase level, electromyography, and muscle biopsy provide more reliable information for diagnosis[5]. Elevation of creatine kinase levels is associated with muscle necrosis, but is not of diagnostic use on its own due to low sensitivity[5]. Electromyography, however, is an essential method in making a fi nal or differential diagnosis. A reduction in the amplitude of compound muscle action potentials or sensory nerve action potentials, or both, with preserved conduction velocity and normal distal motor latencies, in addition to normal responses to repetitive nerve stimulation, may be seen within 2–5 days after the onset of symptoms[57]. The reduction in amplitude often precedes clinical presentation and accompanying fi brillation potentials and positive sharp waves that may occur in the second or third week[15,58-59]. The duration of compound muscle action potentials is an important indicator of critical illness myopathy, and it accompanies the fall in amplitude[11,19,60]. Compound muscle action potential duration can be 2–3 times longer compared with healthy controls, and is most pronounced in lower limb nerves. Electrophysiological screening showing compound muscle action potential duration reductions below two standard deviations of the normal value accurately identifies patients with critical illness polyneuropathy and myopathy[61]. Patients admitted for sepsis/systemic inflammatory response syndrome showing electromyographic signs of characteristic compound muscle action potentials in early stages, and an abnormal baseline nerve conduction study are more likley to develop acquired neuromuscular dysfunction with increased hospital mortality[62]. Baum et al.[63]identi fi ed four different clusters of electrophysiological impairment in patients with sepsis/systemic inflammatory response syndrome, which enabled further differentiation of the severity of neuromuscular disturbances in sepsis-associated organ failure (Cluster 1: normal values for compound muscle action potentials, sensory nerve action potential, and nerve conduction velocity in all nerves, which accounted for 10% of total patients; Cluster 2: pathological values for compound muscle action potentials in the lower extremities and other parameters normal, which accounted for 17% of patients; Cluster 3: moderate pathological values for compound muscle action potentials, sensory nerve action potential, and sensory nerve conduction velocity in upper and lower extremities and motor nerve conduction velocity in lower extremities, which accounted for 40% of patients; Cluster 4: severe disturbances of compound muscle action potentials, sensory nerve action potential, and nerve conduction velocity in upper and lower extremities, which accounted for 33% of patients).
Varying degrees of fibrillation potentials and positive sharp waves can be recorded in both critical illness polyneuropathy and myopathy. Direct muscle stimulation is able to distinguish neuropathy from myopathy[24,64-65]. Critical illness myopathy is characterized by signi fi cant slowing of the muscle fi ber conduction velocity, or even muscle fi ber conduction block during the acute phase, which correlates with prolonged compound muscle action potentials duration and altered muscle fiber excitability[35]. Abnormal sensory nerve action potentials are characteristic for critical illness polyneuropathy, although local edema may interfere with optimal sensory nerve stimulation and recording[19,63,65-67].
Muscle biopsy is still considered the gold standard for confi rming muscle involvement in the disease process despite its invasiveness[11,21]. Critical illness myopathy is pathologically classified into five subtypes: (1) thick filament myopathy; (2) acute myopathy with scattered necrosis; (3) acute myopathy with diffuse necrosis; (4) disuse cachectic myopathy; and (5) rhabdomyolysis (Figure 2)[13,60]. Although unusual, myopathy can progress to frank rhabdomyolysis[13,68-69]. Critical illness polyneuropathy presents as morphological signs of axonal degeneration in both type 1 and type 2 fibers, resulting in extensive denervation atrophy of muscles[57].During recovery, muscle biopsy may show grouped atrophy of muscle fi bers[57]. Furthermore, angular atrophy of isolated scattered muscle fi bers has been observed as part of an acute denervation process[37,46,57,70-71].
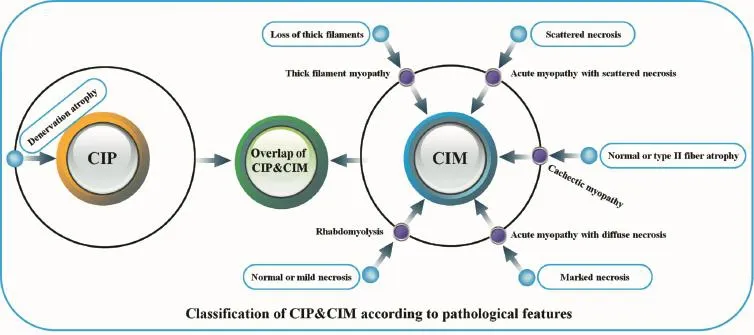
Figure 2 Pathological classi fi cation of critical illness polyneuropathy (CIP) and myopathy (CIM).
Clinical characteristics and examination results should be carefully evaluated for correct diagnosis of critical illness polyneuropathy and myopathy[2,13,46,70-71]. A de fi nite diagnosis of critical illness polyneuropathy requires that the following criteria be met: (1) The critically ill patient develops limb weakness or dif fi culty in weaning, after non-neuromuscular causes such as heart and lung diseases have been excluded; (2) electromyography shows axonal motor and sensory polyneuropathy; (3) absent decremental response to repetitive nerve stimulation[13]. A definite diagnosis of critical illness myopathy requires that the following criteria be met: (1) The critically ill patient develops limb weakness or dif fi culty weaning, after non-neuromuscular causes such as heart and lung diseases have been excluded; (2) compound muscle action potential amplitudes are less than 80% of the normal lower limit in two or more nerves without conduction block; (3) sensory nerve action potential amplitudes are greater than 80% of the normal lower limit; (4) needle electromyography shows short duration, low-amplitude motor unit potentials with early or normal full recruitment, with or without fi brillation potentials, in conscious and collaborative patients, or increased compound muscle action potential duration or reduced muscle membrane excitability on direct muscle stimulation in non-collaborative patients; (5) absent decremental response to repetitive nerve stimulation; (6) muscle histopathology shows primary myopathy[13]. Subsequently, a diagnostic fl owchart for critical illness polyneuropathy and myopathy has been proposed by Latronico and Bolton[13].
Electromyography and nerve conduction studies are theoretically sufficient for clinical diagnosis[72]. However, electrophysiological studies are difficult to conduct in intensive care units due to the presence of edema, inadequate voluntary muscle contraction, and electrical interference[73]. Thus, electrophysiological diagnosis of critical illness polyneuropathy and myopathy is often only possible when patients are fully cooperative, and when nerve conduction studies are performed without interference[73]. Additionally, coexistence of critical illness polyneuropathy and myopathy may make interpretation dif fi cult. Thus, electrophysiological fi ndings should always be correlated with clinical fi ndings. In dif fi cult cases, muscle biopsy may be required for a definite diagnosis. Recently, Tzanis et al.[74]have proposed that maximum inspiratory pressure may serve as a surrogate indicator for the assessment of critical illness polyneuropathy and myopathy. The authors measured maximum inspiratory pressure using the unidirectional valve method, independent of the patient’s ability to cooperate. A signi ficant correlation was found between maximum inspiratory pressure and the Medical Research Council scale score (r = 0.68, P < 0.001)[74]. The fi ndings indicated that maximum inspiratory pressure, estimated using the unidirectional valve method, may be useful for early detection of intensive care unit-acquired weakness[74].
Differential diagnoses
Differential diagnoses of acute neuromuscular weakness commonly include Guillain-Barré syndrome, metabolic neuropathies, toxic neuropathies, and neuropathies due to nutritional de fi ciencies.
Guillain-Barré syndrome, an autoimmune disease of the peripheral nervous system, usually presents as symmetric, progressive, and ascending paralysis, sensory abnormalities and are fl exia, and 30% of patients require mechanical ventilation[75]. The prototype of Guillain-Barré syndrome, which accounts for 90% of all Guillain-Barré syndrome cases in Europe and North America, is acute in fl ammatory demyelin-ating polyneuropathy. The axonal variants of Guillain-Barré syndrome demyelinating polyneuropathy, acute motor axonal neuropathy and acute motor-sensory axonal neuropathy, however, are more prevalent in Asia and South and Central America. The nine most common clinical subtypes of Guillain-Barré syndrome are shown in Table 1.
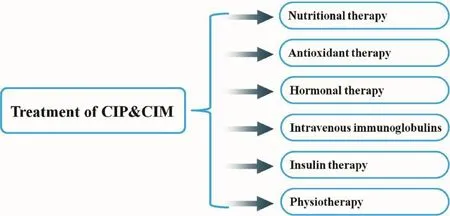
Figure 3 Therapeutic strategies for critical illness polyneuropathy (CIP) and myopathy (CIM).
Different patterns of Guillain-Barré syndrome are probably due to the diverse interaction between antibodies and T cells of divergent specificities[76]. Differentiating critical illness polyneuropathy from Guillain-Barré syndrome, especially the axonal variants, may be dif fi cult on purely clinical grounds, as Guillain-Barré syndrome is known for its variable atypical manifestations[77].
The nerve conduction velocity test shows decreased velocity in typical Guillain-Barré syndrome, whereas normal conduction velocity and decreased action potential, both features of axonal neuropathy, can be seen in critical illness-polyneuropathy and in axonal variants of Guillain-Barré syndrome[58]. The major clinical difference between critical illness polyneuropathy and axonal variants of Guillain-Barré syndrome is that critical illness polyneuropathy is part of a critical illness and it usually occurs during the stay in intensive care, whereas, axonal Guillain-Barré syndrome is a severe form of Guillain-Barré syndrome, which leads to intensive care unit admission[15]. Differentiation between Guillain-Barré syndrome and critical illness polyneuropathy is essential for the determination of therapeutic strategies, as intravenous immunoglobulin or plasma exchange is required in Guillain-Barré syndrome[77], while supportive care is needed in critical illness polyneuropathy[13]. Onset of clinical signs and electrophysiological changes in peripheral nerves and muscles during the development of critical illness polyneuropathy and myopathy can be rapid[13,17,21,78]. Albuminocytologic dissociation in cerebrospinal fluid can help differentiate between the two diseases. The cerebrospinal fluid protein concentrations in patients with Guillain-Barré syndrome are usually normal at the beginning of the disease, and increase in over 90% of patients by the end of the second week[79], whereas protein concentrations are usually unchanged in critical illness polyneuropathy and myopathy. Furthermore, a nerve biopsy and detection of anti-ganglioside antibodies could be helpful in differentiating critical illness polyneuropathy from axonal Guillain-Barré syndrome[15]. The criteria for differentiation are speci fi ed in Table 2[80-81].
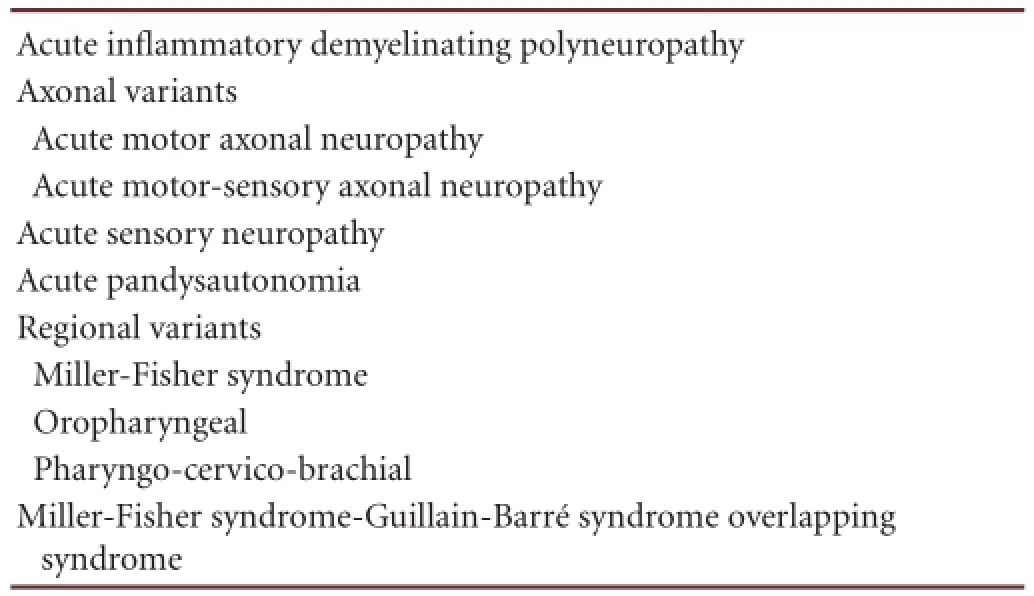
Table 1 Clinical subtypes of Guillain-Barré syndrome
Ionic abnormalities such as hypokalemia, hypophosphatemia, and hypermagnesemia, in addition to the use of several drugs including neuromuscular blocking agents, cancer chemotherapy, statins, and anti-retrovirals, should be differen-tiated because each can affect neuromuscular transmission and cause muscle weakness[13]. Propofol infusion syndrome, a syndrome involving severe metabolic acidosis, cardiac failure, rhabdomyolysis, renal failure, and hypertriglyceridemia after high-dose propofol (5 mg/kg/h) administration for long periods (> 48 hours), is also seen in intensive care units and should be taken into consideration[82]. Propofol infusion syndrome is a rare, but often fatal, syndrome described in critically ill children[82]. Recently, several cases have been reported in adults as well[82]. High-dose propofol, but also supportive treatments using catecholamines and corticosteroids, can act as triggering factors[82].
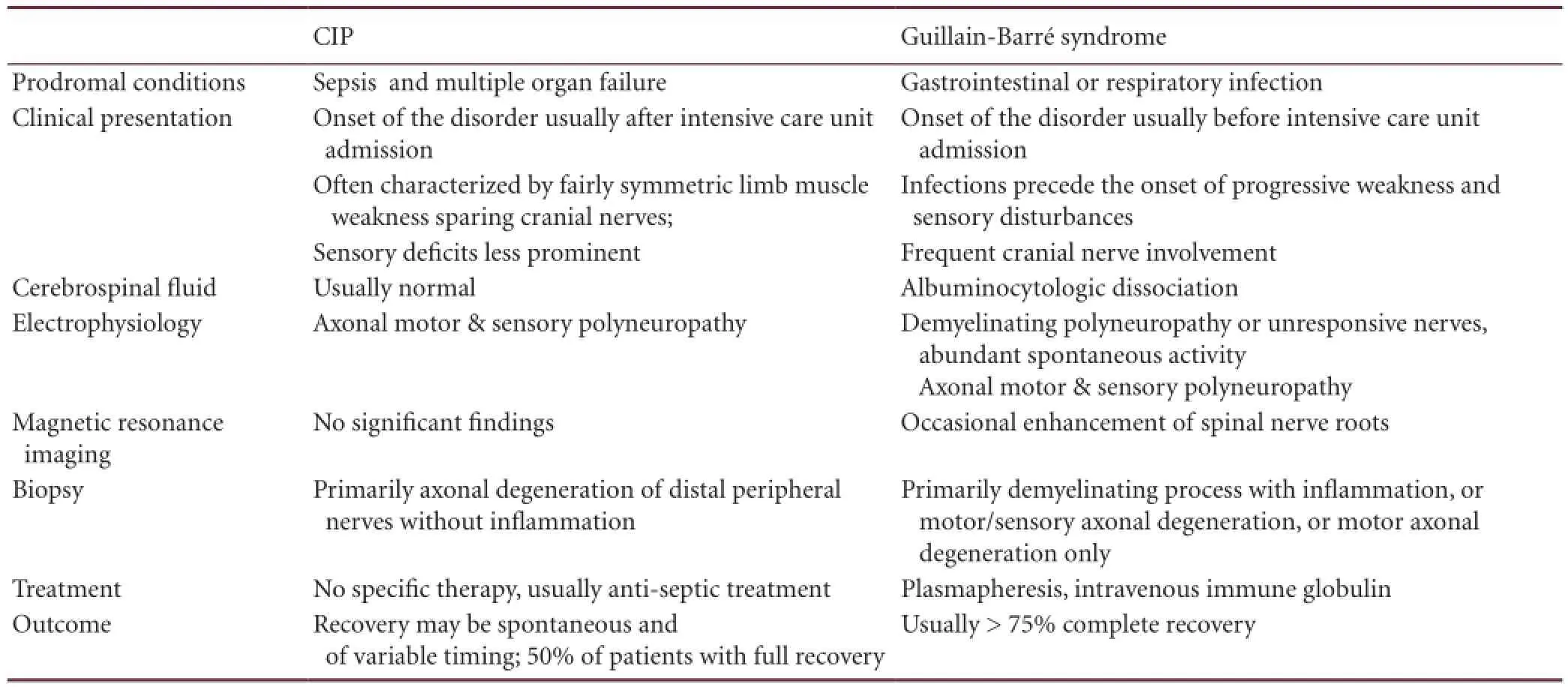
Table 2 Differentiation between critical illness polyneuropathy (CIP) and Guillain-Barré syndrome
Treatment and prognosis of critical illness polyneuropathy and myopathy
Thus far, no speci fi c therapy and only preventive and supportive measures have been shown to be beneficial in the management of critical illness polyneuropathy and myopathy. The measures include aggressive treatment of sepsis, reduction of dose and duration of neuromuscular blocking agents and corticosteroids[36], rehabilitation programs and careful positioning, nutritional interventions[4], anti-oxidant therapy[83], testosterone derivates[84], growth hormones, and immunoglobulins[28,33](Figure 3).
As hyperglycemia is a risk factor for critical illness polyneuropathy and myopathy, strict glycemic control with insulin should have a beneficial effect on patients. Studies have shown that intensive insulin therapy for hyperglycemia lowered the risk of critical illness polyneuropathy and myopathy, and decreased time on ventilatory support, the duration of intensive care unit stay, and the 180-day mortality rate[85-86]. Insulin is associated with some potential beneficial effects, including anti-inflammatory effects[87], endothelial protection[88], improvement of dyslipidemia, and neuroprotective effects in animals[89], and is also an anabolic hormone[5]. A Cochrane systematic review of 825 critically ill patients enrolled in two trials reported that the incidence of critical illness polyneuropathy and/or myopathy was signi fi cantly reduced after intensive insulin therapy[31]. Despite its effectiveness, intensive insulin therapy increases the risk of hypoglycemia. Data from other studies challenged the use of intensive insulin therapy, indicating that strict glycemic control may increase mortality among adults[90]. In a large international randomized trial published in the New England Journal of Medicine, 3,054 critically ill patients were assigned to undergo strict glycemic control (with a target blood glucose range of 4.5–6.0 mmol/L) and 3,050 underwent conventional control (with a target blood glucose range of ≤ 10.0 mmol/L). The investigators found that strict glycemic control increased mortality among adults in intensive care units. Additionally, a blood glucose target of ≤ 10.0 mmol/L resulted in lower mortality compared with a target of 4.5–6.0 mmol/L[90]. Other studies have indicated that the incidence of weakness or failure to wean did not decrease, and that no improvement in muscle strength was achieved by strict glycemic control[76,91].
Early rehabilitation combining mobilization with physiotherapy is emerging as an important strategy to treat critical illness polyneuropathy and myopathy, and to facilitate and improve long-term recovery[24,86,92]and functional independence of patients, and shorten the duration of ventilation and hospitalization[93-94]. Electrical muscle stimulation is a form of exercise and mobilization that does not require patients’ active participation, and can thus be applied to immobilized patients[95]. Electrical muscle stimulation has been shown to be bene fi cial to patients with chronic heart failure[96]and chronic obstructive pulmonary disease[97-98], as well as intensive care unit and hospital inpatients[99-101]. Brie fl y, the protocol of electrical muscle stimulation (45 Hz, 400 μs, 12 seconds on and 6 seconds off, 0.8 second ramp up/ramp down duration) was implemented simultaneously on the vastus lateralis, vastus medialis, and peroneus longus of both lower extremities. Rectangular electrodes wereplaced on the motor points of the aforementioned muscle groups of both legs, and amplitude was set at levels able to cause visible contractions. During the session, the angle of the patients’ knee joint was approximately 40° (0° corresponds to full knee extension). Electrical muscle stimulation sessions lasted for 55 minutes, including 5-minute warm-up and 5-minute recovery periods[95]. A recent study has suggested that electrical muscle stimulation can reduce the loss of muscle mass and exert an acute beneficial effect on the microcirculation, thereby reducing the incidence of critical illness polyneuropathy and myopathy and favorably affecting muscle strength[95]. In a randomized controlled study[95], 142 consecutive patients, with an acute physiology and chronic health evaluation II score of ≥ 13, were randomly assigned to electrical muscle stimulation or control groups. Electrical muscle stimulation was applied daily to the vastus lateralis, vastus medialis, and peroneus longus of both lower extremities. Various muscle groups were evaluated using the Medical Research Council scale for muscle strength[95]. Their results showed that patients in the electrical muscle stimulation group achieved higher Medical Research Council scores compared with controls in wrist fl exion, hip fl exion, knee extension, and ankle dorsiflexion, indicating that the electrical muscle stimulation group performed better in the legs and overall[95]. They concluded that electrical muscle stimulation has bene fi cial effects on the strength of critically ill patients, mainly affecting the muscle groups stimulated, but it may also affect unstimulated muscle groups suggesting a potential effective means of muscle strength preservation and early mobilization in this patient population[95]. Electrical muscle stimulation is bene fi cial to patients who cannot actively exercise, increases muscle strength, and has positive effects on tissue healing and regional vascularization[102-103]. Electrical muscle stimulation is also safe, tolerable, and can be easily applied even in patients unable to cooperate.
Both active and passive therapeutic exercises are recommended as soon as patients are hemodynamically stable, to not only improve neuromuscular function and reduce disability, but also to reduce oxidative stress and in fl ammation[104]. In addition, early mobilization or kinesiotherapy provides faster recovery to function and reduces weaning and hospitalization time[105]. An early mobility and walking program provides guidelines that can assist clinicians who work with patients in intensive care units, especially those receiving mechanical ventilation[105]. The program may facilitate the development of a treatment plan with a focus on individual functional capability, progressive mobilization, and early walking activities. Early mobility in intensive care units can lead to positive outcomes for patients including minimizing complications of bed rest, promoting improved function, promoting weaning from ventilatory support as patients’ overall strength and endurance improves, reducing duration of hospital stay, reducing overall hospital costs, and improving quality of life[105]. Prolonged stays in intensive care units and mechanical ventilation are associated with functional decline and increased morbidity, mortality, cost of care, and length of hospital stay. Implementation of an early mobility and walking program could have a bene fi cial effect on all of the above factors. The program encompasses progressive mobilization and walking procedures, with the progression based on a patient’s functional capability and ability to execute the prescribed activity. The program is divided into four phases. Each phase involves guidelines on positioning, therapeutic exercises, transfers, walking reeducation, and duration and frequency of mobility sessions. The goal in phase 1 is to start mobilization as soon as the patient’s medical condition is stable. Phase 1 includes critically ill patients who are in unstable condition at times and are restricted to bed rest. They can only be out of bed in a stretcher chair because of their inability to bear weight. Phase 2 includes patients whose overall medical condition and muscle strength allow standing activities. These patients progress to transfer training with a walker, prewalking activities, and walking reeducation because of their limited endurance and weakness. Phase 3 involves patients who are ready to start a progressive walking reeducation program and functional training outside the room to improve endurance and functional mobility. Phase 4 involves patients who no longer require ventilatory support and/or have been transferred out of the intensive care unit and are being prepared for hospital discharge[105]. Execution of this program requires a collaborative effort among members of the multi-disciplinary team to coordinate, care for, and provide safe mobilization of patients in intensive care units. It is advisable to initiate rehabilitation as early as possible once diagnosis is established[106-107].
Future therapeutic interventions should be targeted at proinflammatory cytokines, free radical pathways, muscle gene expression, ion channel function, and proteolytic muscle protein mechanisms[108]. The retrospective chart analysis in Mohr et al.[109]suggested that early application of intravenous immunoglobulins may prevent or mitigate critical illness polyneuropathy . However, this awaits con fi rmation in large randomized-controlled studies. Schols and colleagues found that nutritional supplementation in combination with a short course of nandrolone decanoate may enhance the gain in respiratory muscle function in depleted patients with chronic obstructive pulmonary disease, without causing adverse effects[84].
Attention to critical illness polyneuropathy and myopathy outside intensive care units
Critical illness polyneuropathy and myopathy outside the usual setting of the intensive care unit has also attracted attention[110]. There are no precise estimates regarding critical illness polyneuropathy or myopathy outside intensive care units; figures are anecdotal and likely influenced by intensive care unit admission and discharge policies, and hospital resources[110-111]. However, as Latronico and Rasulo[110]proposed, there are two reasons why critical illness polyneuropathy and myopathy outside the intensive care unit merits closer attention. On one hand, critical illness is a functional diagnosis, which is not related to a speci fi c environment. In contrast, the onset of critical illness polyneuropathy and/or myopathy can occur early in critical illness, even before intensive care unit admission. Critical illness polyneuropathy and myopathy are not routinely assessed during the intensivecare unit stay, so the diagnosis of critical illness polyneuropathy and myopathy is limited[110,112]. As a consequence, clinicians may need to treat patients with critical illness polyneuropathy or myopathy after intensive care unit discharge.
Conclusions
In summary, critical illness polyneuropathy and myopathy are frequent complications that occur in patients in intensive care units, especially among those who have acute respiratory distress syndrome, sepsis, systemic in fl ammatory response syndrome, and/or multiple organ failure. This review has attempted to outline the current understanding of critical illness polyneuropathy and myopathy, and underscores the importance of differentiating critical illness polyneuropathy and myopathy from Guillain-Barré syndrome. A guided approach to diagnosis of critical illness polyneuropathy and myopathy is recommended. However, assessment of weakness, polyneuropathy, or myopathy in the intensive care unit setting is sometimes difficult. Presently, the only possible way to reduce the incidence of critical illness polyneuropathy and myopathy is to control risk factors. Both preventive and supportive therapies may be bene fi cial. Avoiding or limiting the use of neuromuscular blocking agents and corticosteroids, as well as early rehabilitation combining mobilization with physiotherapy is crucial to patients’ prognosis. Further investigations are needed to explore effective preventive, diagnostic, and therapeutic strategies for treatment of critical illness polyneuropathy and myopathy.
Author contributions:Wu J and Zhang HL participated in study conception and design. Zhou CK, Wu LM and Ni FM participated in manuscript drafting. Ji W, Wu J and Zhang HL were in charge of manuscript revision for intellectual content. All authors approved the final version of the paper.
Con fl icts of interest:None declared.
Peer review:This study summarized background, definition, pathogenesis, diagnostic criteria and therapy of critical illness polyneuropathy and myopathy, described latest progress of critical illness polyneuropathy and myopathy, and was helpful to understand the nature of nerve injury and regeneration of critical illness polyneuropathy and myopathy.
[1] Lacomis D, Zochodne DW, Bird SJ. Critical illness myopathy. Muscle Nerve. 2000;23(12):1785-1788.
[2] Zochodne DW, Bolton CF, Wells GA, et al. Critical illness polyneuropathy. A complication of sepsis and multiple organ failure. Brain. 1987;110(Pt 4):819-841.
[3] Lacomis D. Neuromuscular disorders in critically ill patients: review and update. J Clin Neuromuscul Dis. 2011;12(4):197-218.
[4] Bolton CF, Laverty DA, Brown JD, et al. Critically ill polyneuropathy: electrophysiological studies and differentiation from Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 1986;49(5): 563-573.
[5] Hermans G, De Jonghe B, Bruyninckx F, et al. Clinical review: Critical illness polyneuropathy and myopathy. Crit Care. 2008;12(6):238.
[6] Gurjar M, Azim A, Baronia AK, et al. Facial nerve involvement in critical illness polyneuropathy. Indian J Anaesth. 2010;54(5):472-474.
[7] Guarneri B, Bertolini G, Latronico N. Long-term outcome in patients with critical illness myopathy or neuropathy: the Italian multicentre CRIMYNE study. J Neurol Neurosurg Psychiatry. 2008;79(7):838-841.
[8] Osler W, The Principles and Practice of Medicine. New York: D. Appleton & Co. 1892.
[9] Mertens HG. Disseminated neuropathy following coma. On the differentation of socalled toxic neuropathy. Nervenarzt. 1961; 32(1):71-79.
[10] Bolton CF, Gilbert JJ, Hahn AF, et al. Polyneuropathy in critically ill patients. J Neurol Neurosurg Psychiatry. 1984;47(11):1223-1231.
[11] Lacomis D, Giuliani MJ, Van Cott A, et al. Acute myopathy of intensive care: clinical, electromyographic, and pathological aspects. Ann Neurol. 1996;40(4):645-654.
[12] Campellone JV. Clinical approach to neuromuscular weakness in the critically ill patient. J Clin Neuromuscul Dis. 2000;1(3):151-158.
[13] Latronico N, Bolton CF. Critical illness polyneuropathy and myopathy: a major cause of muscle weakness and paralysis. Lancet Neurol. 2011;10(10):931-941.
[14] Bednarik J, Vondracek P, Dusek L, et al. Risk factors for critical illness polyneuromyopathy. J Neurol. 2005;252(3):343-351.
[15] Bercker S, Weber-Carstens S, Deja M, et al. Critical illness polyneuropathy and myopathy in patients with acute respiratory distress syndrome. Crit Care Med. 2005;33(4):711-715.
[16] Witt NJ, Zochodne DW, Bolton CF, et al. Peripheral nerve function in sepsis and multiple organ failure. Chest. 1991;99(1):176-184.
[17] Tennilä A, Salmi T, Pettila V, et al. Early signs of critical illness polyneuropathy in ICU patients with systemic in fl ammatory response syndrome or sepsis. Intensive Care Med. 2000;26(9):1360-1363.
[18] Douglass JA, Tuxen DV, Horne M, et al. Myopathy in severe asthma. Am Rev Respir Dis. 1992;146(2):517-519.
[19] Coakley JH, Nagendran K, Yarwood GD, et al. Patterns of neurophysiological abnormality in prolonged critical illness. Intensive Care Med. 1998;24(8):801-807.
[20] Koch S, Spuler S, Deja M, et al. Critical illness myopathy is frequent: accompanying neuropathy protracts ICU discharge. J Neurol Neurosurg Psychiatry. 2011;82(3):287-293.
[21] Khan J, Harrison TB, Rich MM, et al. Early development of critical illness myopathy and neuropathy in patients with severe sepsis. Neurology. 2006;67(8):1421-1425.
[22] Hund E. Neurological complications of sepsis: critical illness polyneuropathy and myopathy. J Neurol. 2001;248(11):929-934.
[23] De Letter MA, Schmitz PI, Visser LH, et al. Risk factors for the development of polyneuropathy and myopathy in critically ill patients. Crit Care Med. 2001;29(12):2281-2286.
[24] Zink W, Kollmar R, Schwab S. Critical illness polyneuropathy and myopathy in the intensive care unit. Nat Rev Neurol. 2009;5(7): 372-379.
[25] Van den Berghe G, Schoonheydt K, Becx P, et al. Insulin therapy protects the central and peripheral nervous system of intensive care patients. Neurology. 2005;64(8):1348-1353.
[26] Gheith O, Al Otaibi T, Abdelhalim M, et al. Successful management of critical illness polyneuropathy and myopathy in renal transplant recipients. Exp Clin Transplant. 2012;10(1):62-66.
[27] Wilmshurst PT, Treacher DF, Lantos PL, et al. Critical illness poly-neuropathy following severe hyperpyrexia. QJM. 1995;88(5): 351-355.
[28] Garnacho-Montero J, Madrazo-Osuna J, García-Garmendia JL, et al. Critical illness polyneuropathy: risk factors and clinical consequences. A cohort study in septic patients. Intensive Care Med. 2001;27(8):1288-1296.
[29] Dussoix P, Chevrolet JC, Cox J, et al. Secondary neuromyopathies in a resuscitation unit. Réanimation Urgences. 1993;2(1):247-258.
[30] Anastasopoulos D, Kefaliakos A, Michalopoulos A. Is plasma calcium concentration implicated in the development of critical illness polyneuropathy and myopathy? Critical Care. 2011;15(5):R247.
[31] Hermans G, De Jonghe B, Bruyninckx F, et al. Interventions for preventing critical illness polyneuropathy and critical illness myopathy. Cochrane Database Syst Rev. 2009;21(1):CD006832.
[32] Amaya-Villar R, Garnacho-Montero J, Garcia-Garmendi JL, et al. Steroid-induced myopathy in patients intubated due to exacerbation of chronic obstructive pulmonary disease. Intensive Care Med. 2005;31(1):157-161.
[33] Boles JM, Bion J, Connors A, et al. Weaning from mechanical ventilation. Eur Respir J. 2007;29(5):1033-1056.
[34] Lacomis DJ. Neuromuscular disorders in critically ill patients: review and update. J Clin Neuromuscul Dis. 2011;12(4):197-218.
[35] Friedrich O, Hund E, Weber C, et al. Critical illness myopathy serum fractions affect membrane excitability and intracellular calcium release in mammalian skeletal muscle. J Neurol. 2004;251(1): 53-65.
[36] Friedrich O. Critical illness myopathy: sepsis-mediated failure of the peripheral nervous system. Eur J Anaesthesiol. 2008;42(1):73-82.
[37] Fenzi F, Latronico N, Refatti N, et al. Enhanced expression of e-selectin on the vascular endothelium of peripheral nerve in critically ill patients with neuromuscular disorders. Acta Neuropathol. 2003;106(1):75-82.
[38] Latronico N, Peli E, Botteri M. Critical illness myopathy and neuropathy. Curr Opin Crit Care. 2005;11(2):126-132.
[39] Gamrin L, Andersson K, Hultman E, et al. Longitudinal changes of biochemical parameters in muscle during critical illness. Metabolism. 1997;46(7):756-762.
[40] Showalter CJ, Engel AG. Acute quadriplegic myopathy: analysis of myosin isoforms and evidence for calpain-mediated proteolysis. Muscle Nerve. 1997;20(3):316-322.
[41] Tiao G, Hobler S, Wang JJ, et al. Sepsis is associated with increased mRNAs of the ubiquitin-proteasome proteolytic pathway in human skeletal muscle. J Clin Invest. 1997;99(2):163-168.
[42] Klaude M, Fredriksson K, Tjäder I, et al. Proteasome proteolytic activity in skeletal muscle is increased in patients with sepsis. Clin Sci (Lond). 2007;112(9):499-506.
[43] Di Giovanni S, Molon A, Broccolini A, et al. Constitutive activation of MAPK cascade in acute quadriplegic myopathy. Ann Neurol. 2004;55(2):195-206.
[44] Di Giovanni S, Mirabella M, D’Amico A, et al. Apoptotic features accompany acute quadriplegic myopathy. Neurology. 2000;55(6): 854-858.
[45] Wagenmakers AJ. Muscle function in critically ill patients. Clin Nutr. 2001;20(5):451-454.
[46] Bolton CF. Neuromuscular manifestations of critical illness. Muscle Nerve. 2005;32(2):140-163.
[47] Rich MM, Pinter MJ. Crucial role of sodium channel fast inactivation in muscle fi bre inexcitability in a rat model of critical illness myopathy. J Physiol. 2003;547(Pt 2):555-566.
[48] Filatov GN, Rich MM. Hyperpolarized shifts in the voltage dependence of fast inactivation of Nav1.4 and Nav1.5 in a rat model of critical illness myopathy. J Physiol. 2004;559(Pt 3):813-820.
[49] Rossignol B, Gueret G, Pennec JP, et al. Effects of chronic sepsis on the voltage-gated sodium channel in isolated rat muscle fi bers. Crit Care Med. 2007;35(2):351-357.
[50] Allen DC, Arunachalam R, Mills KR. Critical illness myopathy: further evidence from muscle- fi ber excitability studies of an acquired channelopathy. Muscle Nerve. 2008;37(1):14-22.
[51] Haeseler G, Foadi N, Wiegand E, et al. Endotoxin reduces availability of voltage-gated human skeletal muscle sodium channels at depolarized membrane potentials. Crit Care Med. 2008;36:1239-1247.
[52] Z’Graggen WJ, Lin CS, Howard RS, et al. Nerve excitability changes in critical illness polyneuropathy. Brain. 2006;129(Pt 9):2461-2470.
[53] Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360(3928):219-223.
[54] Druschky A, Herkert M, Radespiel-Tröger M, et al. Critical illness polyneuropathy: Clinical findings and cell culture assay of neurotoxicity assessed by a prospective study. Intensive Care Med. 2001;27(4):686-693.
[55] Latronico N, Guarneri B. Critical illness myopathy and neuropathy. Minerva Anestesiol. 2008;74(6):319-323.
[56] De Jonghe B, Sharshar T, Lefaucheur JP, et al. Paresis acquired in the intensive care unit: a prospective multicenter study. JAMA. 2002;288(22):2859-2867.
[57] Latronico N, Fenzi F, Recupero D, et al. Critical illness myopathy and neuropathy. Lancet. 1996;347(1):1579-1582.
[58] Bednarik J, Lukas Z, Vondracek P. Critical illness polyneuromyopathy: the electrophysiological components of a complex entity. Intensive Care Med. 2003;29(9):1505-1514.
[59] Cankayali I, Dogan YH, Solak I, et al. Neuromuscular deterioration in the early stage of sepsis in rats. Crit Care. 2007;11(1):R1.
[60] Stibler H, Edström L, Ahlbeck K, et al. Electrophoretic determination of the myosin/actin ratio in the diagnosis of critical illness myopathy. Intensive Care Med. 2003;29(9):1515-1527.
[61] Latronico N, Bertolini G, Guarneri B, et al. Simpli fi ed electrophysiological evaluation of peripheral nerves in critically ill patients: the Italian multi-centre CRIMYNE study. Crit Care. 2007;11(1):R11.
[62] Guarneri B, Bertolini G, Latronico N. Long-term outcome in patients with critical illness myopathy or neuropathy: the Italian multicentre CRIMYNE study. J Neurol Neurosurg Psychiatry. 2008;79(7):838-841.
[63] Baum P, Bercker S, Villmann T, et al. Critical illness myopathy and neuropathy (CRIMYN). Electroneurographic classi fi cation. Nervenarzt. 2011;82(4):468-474.
[64] Rich MM, Raps EC, Bird SJ. Distinction between acute myopathy syndrome and critical illness polyneuropathy. Mayo Clin Proc. 1995;70(2):198-200.
[65] Trojaborg W. Electrophysiologic techniques in critical illness-associated weakness. J Neurol Sci. 2006;242(1-2):83-85.
[66] Rich MM, Bird SJ, Raps EC, et al. Direct muscle stimulation in acute quadriplegic myopathy. Muscle Nerve. 1997;20(6):665-673.
[67] Lefaucheur JP, Nordine T, Rodriguez P, et al. Origin of ICU acquired paresis determined by direct muscle stimulation. J Neurol Neurosurg Psychiatry. 2006;77(4):500-506.
[68] Pandit L, Agrawal A. Neuromuscular disorders in critical illness. Clin Neurol Neurosurg. 2006;108(7):621-627.
[69] Barohn RJ, Jackson CE, Rogers SJ, et al. Prolonged paralysis due to nondepolarizing neuromuscular blocking agents and corticosteroids. Muscle Nerve. 1994;17(6):647-654.
[70] De Letter MA, van Doorn PA, Savelkoul HF, et al. Critical illness polyneuropathy and myopathy (CiPNM): evidence for local immune activation by cytokine expression in the muscle tissue. J Neuroimmunol. 2000;106(1-2):206-213.
[71] Stevens RD, Marshall SA, Cornblath DR, et al. A framework for diagnosing and classifying intensive care unit-acquired weakness. Crit Care Med. 2009;37(10):S299-308.
[72] Wiles CM. Neurological complications of severe illness and prolonged mechanical ventilation. Thorax. 1996;51(Suppl 2):S40-44.
[73] Wieske L, Harmsen RE, Schultz MJ, et al. Is critical illness neuromyopathy and duration of mechanical ventilation decreased by strict glucose control? Neurocrit Care. 2011;14(3):475-481.
[74] Tzanis G, Vasileiadis I, Zervakis D, et al. Maximum inspiratory pressure, a surrogate parameter for the assessment of ICU-acquired weakness. BMC Anesthesiol. 2011;11(1):14.
[75] Rooney KA, Thomas NJ. Severe pulmonary hypertension associated with the acute motor sensory axonal neuropathy subtype of Guillain-Barré syndrome. Pediatr Crit Care Med. 2010;11(1):e16-19.
[76] Hughes RA, Hadden RD, Gregson NA, et al. Pathogenesis of Guillain-Barré syndrome. J Neuroimmunol. 1999;100(1-2):74-97.
[77] Van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol. 2008; 7(10):939-950.
[78] Latronico N, Rasulo FA, Recupero D, et al. Acute quadriplegia with delayed onset and rapid recovery. Case report. J Neurosurg. 1998; 88(4):769-772.
[79] Van der Meché FG, van Doorn PA. Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy: immune mechanisms and update on current therapies. Ann Neurol. 1995; 37(suppl 1):S14-31.
[80] Algahtani H, Moulin DE, Bolton CF, et al. Guillain-Barre syndrome following cardiac surgery. Dif fi cult diagnosis in the intensive care unit. Neurosciences (Riyadh). 2009;14(4):374-378.
[81] Green DM. Weakness in the ICU: Guillain-Barré syndrome, myasthenia gravis, and critical illness polyneuropathy/myopathy. Neurologist. 2005;11(6):338-347.
[82] Vasile B, Rasulo F, Candiani A, et al. The pathophysiology of propofol infusion syndrome: a simple name for a complex syndrome. Intensive Care Med. 2003;29(9):1417-1425.
[83] Yu YM, Ryan CM, Fei ZW, et al. Plasma L-5-oxoproline kinetics and whole blood glutathione synthesis rates in severely burned adult humans. Am J Physiol Endocrinol Metab. 2002;282(2):E247-258.
[84] Schols AM, Soeters PB, Mostert R, et al. Physiologic effects of nutritional support and anabolic steroids in patients with chronic obstructive pulmonary disease. A placebo-controlled randomized trial. Am J Resp Crit Care Med. 1995;152(4 Pt 1):1268-1274.
[85] Morris PE, Goad A, Thompson C, et al. Early intensive care unit mobility therapy in the treatment of acute respiratory failure. Crit Care Med. 2008;36(8):2238-2243.
[86] Hermans G, Wilmer A, Meersseman W, et al. Impact of intensive insulin therapy on neuromuscular complications and ventilator dependency in the medical intensive care unit. Am J Resp Crit Care Med. 2007;175(5):480-489.
[87] Hansen TK, Thiel S, Wouters PJ, et al. Intensive insulin therapy exerts antiin fl ammatory effects in critically ill patients and counteracts the adverse effect of low mannose-binding lectin levels. J Clin Endocrinol Metab. 2003;88(3):1082-1088.
[88] Langouche L, Vanhorebeek I, Vlasselaers D, et al. Intensive insulin therapy protects the endothelium of critically ill patients. J Clin Invest. 2005;115(8):2277-2286.
[89] Ishii DN, Lupien SB. Insulin-like growth factors protect against diabetic neuropathy: effects on sensory nerve regeneration in rats. J Neurosci Res. 1995;40(1):138-144.
[90] NICE-SUGAR Study Investigators, Finfer S, Chittock DR, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360(13):1283-1297.
[91] Hermans G, Schrooten M, Van Damme P, et al. Bene fi ts of intensive insulin therapy on neuromuscular complications in routine daily critical care practice: a retrospective study. Crit Care. 2009; 13(1):R5.
[92] Rochester CL. Rehabilitation in the intensive care unit. Semin Respir and Crit Care Med. 2009;30(6):656-669.
[93] Needham DM. Mobilizing patients in the intensive care unit: improving neuromuscular weakness and physical function. JAMA. 2008;300(14):1685-1690.
[94] Schweickert WD, Pohlman MC, Pohlman AS, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet. 2009; 373(9678):1874-1882.
[95]Karatzanos E, Gerovasili V, Zervakis D, et al. Electrical muscle stimulation: an effective form of exercise and early mobilization to preserve muscle strength in critically ill patients. Crit Care Res Prac. 2012;2012(1):432752.
[96]Nuhr MJ, Pette D, Berger R, et al. Bene fi cial effects of chronic low-frequency stimulation of thigh muscles in patients with advanced chronic heart failure. Eur Heart J. 2004;25(2):136-143.
[97]Vivodtzev I, Pépin JL, Vottero G, et al. Improvement in quadriceps strenght and dyspnea in daily tasks after 1 month of electrical stimulation in severely deconditioned and malnourished COPD. Chest. 2006;129(6):1540-1548.
[98]Sillen MJ, Speksnijder CM, Eterman RM, et al. Effects of neuromuscular electrical stimulation of muscles of ambulation in patients with chronic heart failure or COPD: a systematic review of the English-language literature. Chest. 2009;136(1):44-61.
[99]Gruther W, Kainberger F, Fialka-Moser V, et al. Effects of neuromuscular electrical stimulation on muscle layer thickness of knee extensor muscles in intensive care unit patients: a pilot study. J Rehabil Med. 2010;42(6):593-597.
[100]Strasser EM, Stättner S, Karner J, et al. Neuromuscular electrical stimulation reduces skeletal muscle protein degradation and stimulates insulin-like growth factors in an age- and current-dependent manner: a randomized, controlled clinical trial in major abdominal surgical patients. Ann Surg. 2009;249():738-743.
[101]Bouletreau P, Patricot MC, Saudin F, et al. Effects of intermittent electrical stimulations on muscle catabolism in intensive care patients. JPEN J Parenter Enteral Nutr. 1987;11(6):552-555. [102]Routsi C, Gerovasili V, Vasileiadis I, et al. Electrical muscle stimulation prevents critical illness polyneuromyopathy: a randomized parallel intervention trial. Crit Care. 2010;14(2):R74.
[103]Pattanshetty RB, Gaude GS. Critical illness myopathy and polyneuropathy - A challenge for physiotherapists in the intensive care units. Indian J Crit Care Med. 2011;15(2):78-81.
[104]Silva A, Maynard K, Cruz M. Effect of motor physical therapy in critically ill patients. Rev Bras Ter Intensiva. 2010;22(1):85-91.
[105]Perme C, and Chadrashekhar R. Early mobility and walking program for patients in intensive care units: creating a standard of care. Am Crit Care. 2009;18(1):212-221.
[106]Novak P, Vidmar G, Kuret Z, et al. Rehabilitation of critical illness polyneuropathy and myopathy patients: an observational study. Int J Rehabil Res. 2011;34(4):336-342.
[107]Kress JP. Clinical trials of early mobilization of critically ill patients. Crit Care Med. 2009;37(10 Suppl):S442-447.
[108]Chawla J, Gruener G. Management of critical illness polyneuropathy and myopathy. Neurol Clin. 2010;28(4):961-977.
[109]Mohr M, Englisch L, Roth A, et al. Effects of early treatment with immunoglobulin on critical illness polyneuropathy following multiple organ failure and gram-negative sepsis. Intensive Care Med. 1997;23(11):1144-1149.
[110]Latronico N, Rasulo FA. Presentation and management of ICU myopathy and neuropathy. Curr Opin Crit Care. 2010;16(2):123-127.
[111]Latronico N, Guarneri B, Alongi S, et al. Acute neuromuscular respiratory failure after ICU discharge. Report of fi ve patients. Intensive Care Med. 1999;25(11):1302-1306.
[112]Latronico I, Shehu I, Guarneri B. Use of electrophysiologic testing. Crit Care Med. 2009;37(10 Suppl):S316-320.
Copyedited by Barrett R, Yajima W, Qi XK, Duan RS, Wang LM, Qiu Y, Li CH, Song LP, Liu WJ, Zhao M
10.4103/1673-5374.125337
Hongliang Zhang, M.D., Ph.D., Department of Neurology, the First Bethune Hospital, Jilin University, Xinmin street 71#, Changchun 130021, Jilin Province, China, drzhl@hotmail.com, Hongliang. Zhang@ki.se.
http://www.nrronline.org/
Accepted: 2013-11-25
- 中国神经再生研究(英文版)的其它文章
- Lipid rafts participate in aberrant degradative autophagic-lysosomal pathway of amyloid-beta peptide in Alzheimer’s disease
- Electro-acupuncture at Conception and Governor vessels and transplantation of umbilical cord bloodderived mesenchymal stem cells for treating cerebral ischemia/reperfusion injury
- Hippocampal gene expression in a rat model of depression after electroacupuncture at the Baihui and Yintang acupoints
- A non-invasive, rapid method to genotype late-onset Alzheimer’s disease-related apolipoprotein E gene polymorphisms
- Mechanism underlying the protective effect of Kaixin Jieyu Fang on vascular depression following cerebral white matter damage
- Changes in brain functional network connectivity after stroke