
Heterologous expression, chaperone mediated solubilization and purification of parasitic nematode-specific growth factor-like protein of Setaria digitata
2014-03-22 09:16WPRodrigoDassanayakeKarunanayakeSilvaGunawardeneVDSWeerasena
W. WP. Rodrigo, R. S Dassanayake, E. H. Karunanayake, Y.I.N. Silva Gunawardene, O. VDS. J. Weerasena
1Department of Chemistry, Faculty of Science, University of Colombo, 90, Cumaratunga Munidasa Mawatha, Colombo 03, Sri Lanka
2Institute of Biochemistry, Molecular Biology and Biotechnology, University of Colombo, 90, Cumaratunga Munidasa Mawatha, Colombo 03, Sri Lanka
3Molecular Medicine Unit, Faculty of Medicine, University of Kelaniya, Ragama, Sri Lanka
Heterologous expression, chaperone mediated solubilization and purification of parasitic nematode-specific growth factor-like protein of Setaria digitata
W. WP. Rodrigo1,2, R. S Dassanayake1*, E. H. Karunanayake2, Y.I.N. Silva Gunawardene3, O. VDS. J. Weerasena2
1Department of Chemistry, Faculty of Science, University of Colombo, 90, Cumaratunga Munidasa Mawatha, Colombo 03, Sri Lanka
2Institute of Biochemistry, Molecular Biology and Biotechnology, University of Colombo, 90, Cumaratunga Munidasa Mawatha, Colombo 03, Sri Lanka
3Molecular Medicine Unit, Faculty of Medicine, University of Kelaniya, Ragama, Sri Lanka
Objective: To clone, express and purify a putative parasitic nematode specific protein of Setaria digitata (S. digitata), filarial nematode that infects livestock and cause significant economic losses in Far East and Asia to be used for structural and functional analyses.
Methods: To characterize uncharacterized gene of S. digitata (SDUG), the herterologous expression of SDUG was carried out in the pET [cloned into pET45b(+)] expression system initially and co-expression of SDUG using chaperone plasmids pG-KJE8, pGro 7, pKJE7, pG-Tf2 and pTf16 containing chaperone proteins of dnaK-dnaJ-grpE-groES-gro-E, groES-groEL, dnaK-dnaJ-grpE, groES-groEL-tig, and tig respectively, was carried out subsequently. Results: Expression of SDUG was seen when Escherichia coli strain BL21(DE3) is used, while concentrating protein largely into the insoluble fraction. The co-expression of SDUG using chaperone plasmid mediated system indicated a significant increase of the protein in the soluble fraction. Of the chaperon plasmid sets, the highest amount of recombinant SDUP in the soluble fraction was seen when pGro7 was used in the presence of 2 mg/mL L-arabinose and 0.6M IPTG concentration in the culture medium and for 3 h of incubation at the temperature of 28 ℃. Recombinant SDUG was purified both from soluble and insoluble fractions using Ni affinity chromatography. SDS-PAGE and western blot analyses of these proteins revealed a single band having expected size of ~24 kDa. Conclusions: SDUG seems to be more aggregate-prone and hydrophobic in nature and such protein can make soluble by correct selecting the inducer concentrations and induction temperature and its duration.
ARTICLE INFO
Article history:
Received 7 October 2013
Received in revised form 15 November 2013
Accepted 15 January 2014
Available online 20 February 2014
Setaria digitata
1. Introduction
Setaria digitata(S. digitata) is a filarial parasite that is mainly found in the peritoneal cavity of many ungulates and they are nonpathogenic in their natural host cattle and they may cause a mild fibrinous peritonitis for their natural host. The major pathogenic effect occurs when the infective stage larvae (L3) enter into abnormal hosts such as goat, sheep or horses through a mosquito vector that cause cerebrospinal nematodiasis[1]. Further, that this nematode can also infect humans and cause abscesses, allergic reactions, enlarged lymph nodes, eye lesions and lung inflammation showing gradual adaptation to humans. Therefore, this study was undertaken as an initial step towards unraveling the biochemistry of uncharacterized gene of theS. digitatathat has 88% sequence similarity with the human lymphaticfilarial parasites ofWucheretia bancroftiandBrugia malayi (B. malayi ), and 82% sequence similarity to subcutaneous filarial parasite ofLoa loa(the eye worm) and significant sequence similarity with other parasitic nematodes[2]. Bioinformatic analyses of the coding region ofS. digitatauncharacterized gene (SDUG) revealed that the latter protein is a non-enzymatic, rich in beta pleated strands, expressed in all stages in the life cycle ofB. malayi, localized to the nucleus, parasitic nematode specific and growth factor like protein[2].
Escherichia coli(E. coli) is one of the most widely used host for the production of heterologus proteins as it produces large quantities of recombinant proteins in rapid, often inexpensive and high-density cultivation[3]. Major limitation of this system is that the deposition of protein of interest frequently into insoluble inactive aggregates or inclusion bodies[4]. Formation of inclusion bodies sometime may be advantageous in purification of protein because they are easily isolated by centrifugation to yield highly concentrated and relatively pure protein and also latter formation protects the protein from proteolytic attack. In addition, even though if the protein of interest is toxic to the cell, inactive form of the protein as inclusion bodies may not inhibit cell growth[5]. Therefore, this approach is often used for the production of antigens which does not require proper folding. However, inclusion body formation is a major drawback for the structural and/or biochemical studies that requires native proteins. Insufficient availability of molecular chaperones in bacterial system is the major reason for larger fraction of recombinant protein to be localized into inclusion bodies[6]. Therefore, co-expression of chaperones along with foreign protein is a common approach to increase the yield of properly folded recombinant proteins in bacterial systems. Further, co-expression greatly facilitates the analysis of multi-subunit complexes, biochemical pathways, characterization of protein-protein interactions[7] and structural and/or biochemical studies. This paper details the expression of SDUG in pET expression system and purification of recombinant protein in denaturing conditions and also the optimization of plasmids mediated chaperone co-expression approach to obatin protein into soluble fraction for the purification of protein in native form.
2. Methods
2.1. Vector construction for expression of SDUG
Coding sequence of pSDC13 cDNA clone (GenBank Database accession number GU222920.1) from cDNA library ofS. digitatawas used in this study. The cDNA encoding SDUG was amplified by PCR using two primers containing aKpnⅠ (UNFKpn1: 5’ CGG GGT ACC ATG AAT GTG AAA ACA AGG AA 3’) and aXhoⅠ (PICHStXho1: 5’ A TGC ATG CTC GAG TCA TCA GTA ATT AAT CAA ATT CGG AA 3’) sites at N-terminus and C-terminus respectively to clone in pET expression system. Forward primer was designed to inframe with start codon and the N-terminal His-Tag of the pET vector. PCR amplification was performed using 10 ng of plasmid DNA in a 25 μL reaction containing 0.5 unit ofTaqDNA polymerase (GenScript, USA), 160 μM of each dNTP (Promega) and 80 pM of each primers. Reaction was incubated at 95 ℃ for 2 min, followed by 25 cycles of denaturation at 94 ℃ for 30 s, annealing at 50 ℃ for 30 s and extension at 72 ℃ for 1 min, with a final extension step of 72 ℃ for 7 min.
The amplified fragment with UNFKpn1 and PICHStXho1 primers was cloned inKpnⅠ andXhoⅠ sites of the pET45b(+) expression vector (Novagen, USA) and transformed into XL1-Blue MRF competent cells by electrotransformation. Colonies were selected on Luria-Bertani agar (1% tryptone, 0.5% yeast extract, 0.5% NaCl, 1.5% agar) supplemented with ampicillin (50 μg/mL) and the recombinant construct named as pET45b(+)-SDUG (Figure 1) was confirmed by sequencing.

Figure 1. Schematic representation of the recombinant constructs of SDUG in pET expression system.6×His: Codons that code for N-terminal six histidine amino acid residues; SDUG: ORF of SDUG; Stop: Stop codon. Restriction enzymes engineered in primers to clone the recombinant constructs to the vectors [pET 45b(+)] are shown.
2.2. Transformation and screening of SDUG
The recombinant construct of pET45b(+)-SDUG and pET45b(+) vector DNA were transformed intoE. coliBL21 and BL21(DE3) competent cells by electroporation. Transformants were selected on LB agar with ampicillin (50 μg/mL). Recombinant colonies were selected and confirmed by PCR with insert specific primers (UNFKpn1 and PICHStXho1).
2.3. Expression of recombinant SDUG (r-SDUG)
Expression was carried out according to the guidelinesgiven in pET system user manual (Novagen). In brief, overnight cultures were inoculated into 200 mL of LB medium containing 50 μg/mL of ampicillin. Cells were grown at 37 ℃ and induced by addition of either 0.4 mM, 0.6 mM or 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and incubated at the temperatures of either 25 ℃, 28 ℃, 30 ℃ or 33 ℃ for the durations of either 1 h, 3 h, 5 h or 7 h. Before induction, cultures were divided into two and one was used as an un-induced control. Samples were adjusted to the same OD600value for analysis of expression levels and solubility of the target protein by SDS-PAGE.
2.4. SDS-PAGE and Western blot analyses for r- SDUG
The bacterial cells were thawed and sonicated in sodium phosphate buffer (pH 7.4) containing 1 mM PMSF. Samples were centrifuged and the cell lysates and the cell debris were analyzed separately using 10% SDS-PAGE following standard procedures described under Sambrook et al[8]. Samples were mixed with equal volume of 1×SDS gel loading buffer [(62.5 mM Tris HCl (pH 6.8), 10% Glycerol, 2% SDS, 0.01% bromophenol blue, 2.5% β-mercaptoethanol] and boiled for 3 min in a water bath. Then 12 μL of each sample per well was loaded into SDS-PAGE and electrophoresis was performed for the separation of proteins. Bands were visualized by Coomassie blue staining.
Proteins on SDS-PAGE were transferred into nitrocellulose membrane for confirmation of the recombinant protein by western blotting. Western blotting was carried out as the standard method described by Sambrooket al[8] with minor modifications. Nitrocellulose membrane blocked with 5% non fat dry milk for 2 h at 4 ℃ on a platform shaker and incubated for overnight with 2:1 000 anti-His tag antibody (Novagen and MyBioSource). Membrane was washed three times with PBST (1× PBS, 0.05% Tween-20) and incubated immediately with 1:5 000 goat anti-mouse IgG-AP antibodies (Novagen) at 4 ℃ for 2 h. Then the membrane was washed three times with 1× PBS and bands were visualized by using the BCIP/NBT substrate of Biotin Chromogenic Detection Kit (Fermentas). Protein Ladder marker was stained separately with ponceau S staining (Sigma, St. Louis, MO, USA).
2.5. Chaperone mediated co-expression of r-SDUG
2.5.1. Construction of a bacterial Co-expression system to increase soluble fraction of r-SDUG
Five types of chaperone plasmids (pG-KJE8, pGro7, pKJE7, pG-Tf2 and pTf16) were purchased from TaKaRa Biotechnology and transformed them into BL21(DE3) chemical competent cells containing target gene construct of pET45b(+)-SDUG by heat shock method. Transformants were selected on LB containing 20μg/mL chloramphenicol and 50 μg/mL ampicillin.
2.5.2. Co-expression of r-SDUG with chaperone proteins
Five types of chaperones were separately co-expressed with the target gene to select the best chaperon which helps to express highest amount of target protein in soluble fraction. The single colonies of transformants were inoculated separately into LB medium containing 20 μg/mL chloramphenicol and 50 μg/mL ampicillin. Different amount of L-arabinose and tetracycline were added for induction of chaperone expression. L-arabinose and tetracycline were used to induce pG-KJE8, only L-arabinose for pGro7, pKJE7 and pTf16 and only tetracycline for pGTf2 according to the manufacturer’s instructions[9]. Cultures were incubated at 37 ℃ with shaking (250-300 rpm) until OD600reached to 0.4-0.6 and left them at 15 ℃ for 30 min. Then cultures were induced with IPTG to final concentrations of either 0.4 mM, 0.6 mM or 1 mM, and incubated at the temperatures of either 25 ℃, 28 ℃, 30 ℃ or 33 ℃ for the durations of either 1 h, 3 h, 5 h or 7 h. Samples were adjusted to the same OD600value for analysis of expression levels and solubility of the target protein by SDS-PAGE.
2.6. Preparation and purification of r-SDUG by ammonium sulphate method
Cell pellet was dissolved in 1×Ni-NTA denaturing lysis/ binding buffer (8 M urea, 100 mM Sodium Phosphate buffer, 10 mM Tris.HCl, pH 8.0), centrifuged and cell lysate was taken. To determine the optimum precipitation conditions, ammonium sulfate powder was added according to the ammonium sulfate table discuss under the Green and Hughes[10] to adjust solutions to final saturations of 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80% and 90% respectively. Precipitated protein was recovered at each stage by centrifuged at 13 000 rpm for 20 min. Each protein precipitate was dissolved individually in SDS-PAGE loading buffer, boiled, centrifuged and supernatant was run on 10% SDS-PAGE to detect best saturation level for the purification of the recombinant protein from the whole cell lysate.
2.7. Purification of polyhistidine-tagged r-SDUG under denaturing conditions by Ni affinity column
Preparation of bacterial cell lysate and purification of the 6×his-tag protein was performed according to the supplier’s instruction given in the manual of Ni-NTA His-Bind Resins (Novagen). Cell pellet was dissolved in 1×Ni-NTA denaturing lysis/binding buffer (8M urea, 100 mM sodium phosphate buffer, 10 mM Tris.HCl, pH 8.0). Insoluble materials were removed by centrifuging at 12 000 rpm for 30 min and cell lysate was taken carefully without disturbing the pellet. Cell lysate was loaded on the Ni-NTA column, pre-equilibrated with 1× Ni-NTA denaturing lysis/binding buffer. The column was washed with 1×Ni-NTA wash buffer (8 M urea, 100 mM Sodium Phosphate buffer, 10 mM Tris.HCl, 10 mM Imidazole, pH 8.0) and recombinant protein was eluted by minimum volume of 1×Ni-NTA elution buffer (8 M urea, 100 mM Sodium Phosphate buffer, 10 mM Tris.HCl, 250 mM Imidazole, pH 8.0). The size and the purity of the eluted protein were determined by 10% SDS-PAGE and western blotting. Remaining urea and imidazole were removed and concentrated by Amicon® Ultra-15 centrifugal filter devices and the concentration of the protein was determined by Bradford method.
2.8. Preparation and purification of polyhistidine-tagged r-SDUG under native conditions
Preparation of bacterial cell lysate and purification of the 6 ×his-tag protein was performed according to the supplier’s instruction given in the manual of Ni-NTA His-Bind Resins (Novagen). Culture pellet was re-suspended in 1×Ni-NTA bind buffer (300 mM NaCl, 50 mM sodium phosphate buffer, 5 mM imidazole, pH 8.0) in 2-5 mL per gram wet weight. Lysozyme solution was added to a final concentration of 45-60 kU/g. Sample was incubated at 30 ℃ for 15 min and cells were disrupted by sonication having PMSF final concentration of 1 mM. When viscosity was high, RNase (10 μg/mL), DNase Ⅰ (5 μg/mL) and Benzonase® Nuclease (25 U of 1 μL per 1 mL of lysis buffer) were added and incubated on ice for 10-15 min. Samples were centrifuged at 10 000×gfor 20-30 min at 4 ℃ to obtain clear cell lysate. Cell lysate was loaded on the Ni-NTA column, preequilibrated with 1×Ni-NTA binding buffer. The column was washed with 1×Ni-NTA wash buffer (300 mM NaCl, 50 mM sodium phosphate buffer, 20 mM Imidazole, pH 8.0) and recombinant protein was eluted by minimum volume of 1×Ni-NTA elution buffer (300 mM NaCl, 50 mM sodium phosphate buffer, 250 mM imidazole, pH 8.0). Flow-through, Wash fractions and elution fractions were analyzed by SDSPAGE. Protein sample was concentrated by Amicon® Ultra-15 centrifugal filter devices and the concentration was determined by Bradford method.
3. Results
3.1. Cloning and expression of SDUG in E. coli strains BL21 and BL21(DE3)
To express SDUG in bacterial expression system, recombinant clone of pET45b(+)-SDUG (Figure 1) was constructed. pDNA containing the construct identified by PCR amplification using insert specific primers (UNFKpn1 and PICHStXho1) and confirmed by sequencing was used to transformE. coliexpression cells, BL21 and BL21(DE3). The expression of SDUG was carried out using BL21; [BL21-pET45b(+)-SDUG] and BL21(DE3); [BL21(DE3)-pET45b(+)-SDUG] recombinant colonies confirmed by PCR. In this study, theE. colistrains BL21; [BL21-pET45b(+)] and BL21(DE3); [BL21(DE3)-pET45b(+)] transformed by the parent plasmid pET45b(+) were used as vector controls/ negative controls and the induction control strain provided by the manufacturer was used as expression control. SDSPAGE analyses of lysates derived from positive clones revealed an extra band having expected size of 24 kDa in recombinant clone BL21(DE3)-pET45b(+)-SDUG (Figure 2) but not in recombinant clone BL21-pET45b(+)-SDUG following induction with IPTG, when compared with controls indicating expression of r-SDUG in BL21(DE3). Therefore, the recombinant clone BL21(DE3)-pET45b(+)-SDUG was used in subsequent analyses.
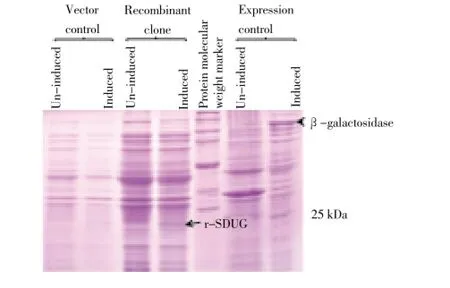
Figure 2. r-SDUG expression in BL21(DE3) strain using SDS-PAGE analysis.Lane (1) & (2), (3) & (4) and (6) & (7) are un-induced and induced samples of strains BL21(DE3)-pET45b(+) , BL21(DE3)-pET45b(+)-SDUG and expression control respectively. Lane (5) broad range protein molecular weight marker (Promega).
3.2. Analysis of r-SDUG in soluble and insoluble fractions
To devise an appropriate purification strategy, a study was carried out to investigate the relative distribution of the expressed recombinant protein of clone BL21(DE3)-pET45b(+)-SDUG in the soluble and insoluble fractions. Both the cell lysate (soluble fraction) and the cell debris (insoluble fraction) were examined after sonication in 1X PBS for the expression of r-SDUG. The recombinant protein was seen to be present in higher concentration in the insoluble fractions in SDS-PAGE analysis (Figure 3A) in all IPTG concentrations, incubation temperatures and durations of incubation tested. The expression of r-SDUG in BL21(DE3) was confirmed by western blotting with anti-His tag antibody (Figure 3B). As majority of r-SDUG was seen to be concentrated in the insoluble fraction of BL21(DE3), attempts were made to increase soluble r-SDUG by co-expressing SDUG in the presence of chaperone plasmids in obtaining sufficient quantities of correctly folded protein for structural studies.
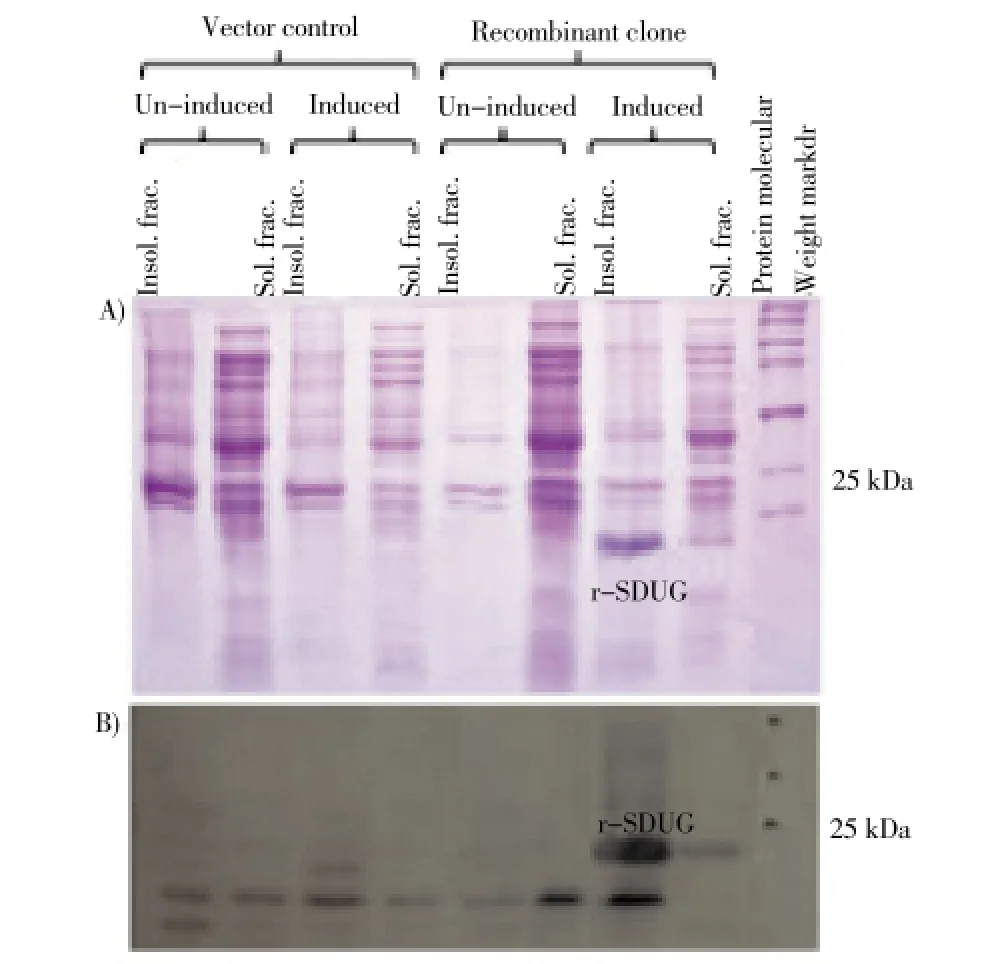
Figure 3. r-SDUG expression in insoluble (cell debris) and soluble fractions (cell lysates) of BL21(DE3) strain using SDS-PAGE (A) and Western blotting (B) analyses.Lane (1) & (4) and (5) & (8) are insoluble (Insol. frac,) and soluble fractions (Sol. frac.) of un-induced and induced samples of vector control strain BL21(DE3)-pET45b(+) and recombinant strain BL21(DE3)-pET45b(+)-SDUG, respectively, and Lane (9) broad range protein molecular weight marker (Promega).
3.3. Chaperone mediated co-expression of r-SDUG
3.3.1. Effect of IPTG concentration and incubation temperature and its duration on r-SDUG expression
The recombinant strain BL21(DE3)-pET45b(+)-SDUG transformed with five types of chaperone team plasmids is used to study the effect of the induction concentration of IPTG, temperature of incubation, duration of incubation on soluble r-SDUG expression. Analysis of cell lysates of bacteria induced with different IPTG concentrations of 0.4 mM, 0.6 mM and 1 mM harvested following 5 h of incubation time revealed the highest degree of soluble r-SDUG expression is taking place in the presence of 0.6 mM of IPTG concentration. Further, analysis of cell lysates of bacteria induced with IPTG concentration of 0.6 mM harvested following 5 h of incubation time at the temperatures of 25, 28, 30 and 33 ℃ revealed the highest level soluble r-SDUG expression is taking place at the temperature of 28 ℃ despite insoluble r-SDUG expression increases with increase of the temperature. Furthermore, analysis of the effect of time of incubation (1 h, 3 h, 5 h and 7 h) on the soluble expression of r-SDUG at the induction IPTG concentration of 0.6 mM and incubation temperature of 28 ℃ revealed the highest level soluble expression r-SDUG is taking place at the 3 h of incubation (Figures 4A & 4B).
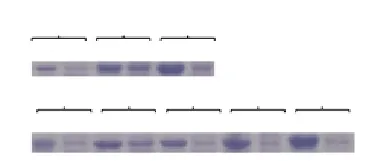
Figure 4. SDS-PAGE analysis to show the effect of induction IPTG concentrations (A) and incubation temperatures (B) for the expression of r-SDUG.Lane (1 & 2), (3 & 4) and (5 & 6) are expression of r-SDUG in insoluble and soluble fractions of bacterial cell lysates at IPTG concentrations of 0.4 mM, 0.6 mM and 1 mM respectively at 28 ℃for 3 h incubation. Lane (7 & 8), (9& 10), (11&12), (13&14) and (15 &16) are expression of r-SDUG in insoluble and soluble fractions of bacterial lysates at temperatures of 25 ℃, 28 ℃, 30 ℃, 33 ℃ and 37 ℃ respectively at 0.6 mM induction IPTG concentration for 3 h incubation.
3.3.2. Effect of L-arabinose and tetracycline on r-SDUG expression
SDUG was co-expressed separately with chaperoneplasmids. Different concentrations of L-arabinose and tetracycline were used to induce expression of chaperones to ascertain which concentration of L-arabinose and tetracycline and which type of chaperone plasmid was the best for the expression of target gene in soluble fraction. The results indicated the chaperone plasmid pGro7 produces the highest amount of recombinant protein in the soluble fraction in the presence of 2 mg/mL L-arabinose (Figure 5). Therefore, BL21(DE3)-pET45b(+)-SDUG transformed with chaperone plasmid pGro7 together with the appropriate amount of L-arabinose were used for bacterial recombinant protein expression in the presence of IPTG concentration of 0.6 mM, incubation temperature of 28 ℃ and the incubation duration of 3 h.
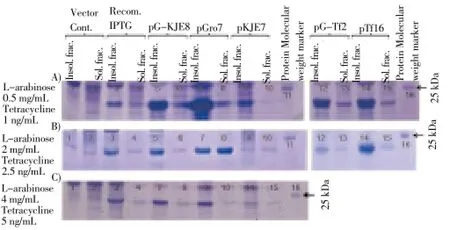
Figure 5. Comparison of co-expression of r-SDUG in strain BL21(DE3) with five types of chaperone plasmids with the induction of different concentrations of L-arabinose and tetracycline.Lane (1) & (2) and (3) & (4) are insoluble and soluble fractions of vector control [BL21(DE3)-pET45b(+)] and recombinant clone [BL21(DE3)-pET45b(+)-SDUG], respectively. Lane (5) & (6), (7) & (8), (9) & (10), (12) & (13) and (14) & (15) are insoluble and soluble fractions of BL21(DE3) co-expressed with pG-KJE8, pGro7, pKJE7, pG-Tf2 and pTf16, respectively, induced with different concentrations of L-arabinose and Tetracycline [0.5 mg and 1 ng/mL (A), 2.0 mg and 2.5 ng/mL (B) & 4 mg and 5 ng/mL (C)] and followed by IPTG induction concentration of 0.6 mM, at 28 ℃ for 3 h and Lane (11) & (16) broad range protein molecular weight marker (Promega).
3.4. Purification of r-SDUG
Ammonium sulfate precipitation was carried out to concentrate and to purify the recombinant soluble SDUG expressed in the clone BL21(DE3)-pET45b(+)-SDUG. SDSPAGE analysis revealed that r-SDUG precipitate in all ammonium sulfate saturation levels in different quantities. However, the higher level of r-SDUG precipitation was seen in the range of 20% to 40% of ammonium sulfate saturation level with some degree of impurities (Figure 6). Therefore, this method was not adopted in the purification; instead, Ni-NTA column chromatography was used. His-tagged r-SDUG was purified in denaturing conditions from insoluble fraction and also native conditions from soluble fraction using latter chromatographic technique and both the SDS-PAGE analysis (Figure 7A) and western blot analysis (Figure 7B) following reaction with an anti-His antibody detected a single band of 24 kD corresponding to the recombinant protein.
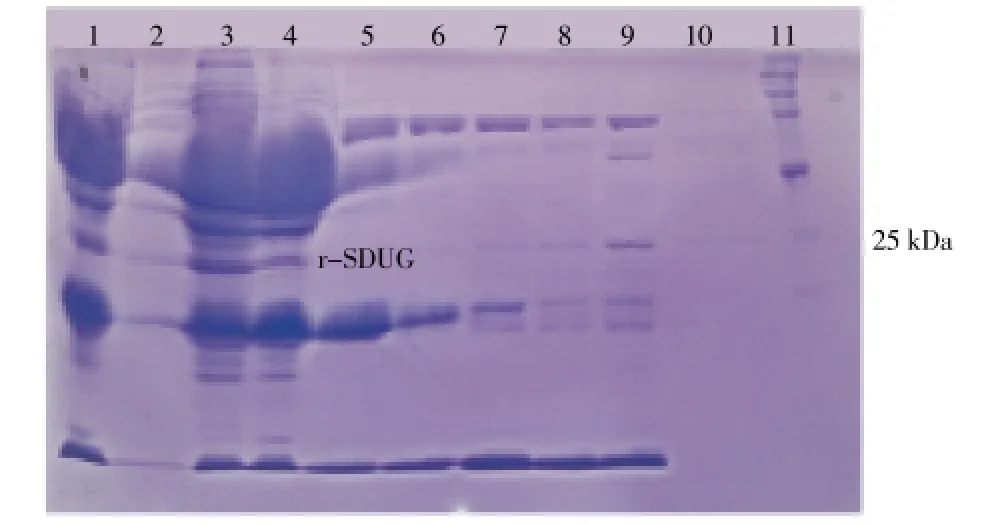
Figure 6. SDS-PAGE analysis of r-SDUG precipitated at different concentration of ammonium sulfate from cell lysate of strain BL21(DE3).Lane (1) Whole cell lysate; Lane (2)-(10) the ammonium sulfate saturation of 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80% and 90% respectively, and Lane (11) broad range protein molecular weight marker (Promega).

Figure 7. SDS-PAGE (A) and Western blotting (B) analyses of purified r-SDUG from cell lysates of strain BL21(DE3) using highaffinity Ni-IDA column chromatography.Lane (1) whole cell lysate, Lane (2-3) concentrated Elution fractions and Lane (M) broad range protein molecular weight marker (Promega).
4. Discussion
This study details the expression and purification of a parasitic nematode-specific protein of the cattle filarial parasiteS. digiatausing the pET expression systems. To express SDUG in bacteria, the pET system was used. pET system is a powerful T7 RNA polymerase drivenE. colisystem which can express more than 50% of recombinant protein of the total cell proteins within a few hours followinginduction[11]. SDUG obtained from the cDNA clone of pSDC13 was cloned into pET expression vector pET45b(+) and transformed into BL21 and BL21(DE3)E. colistrains for expression. Level of protein expression was analyzed on SDS-PAGE with the cell lysates obtained from bothE. colistrains transformed with pET45b(+)-SDUG constructs following induction with IPTG. Induced and uninduced samples were analysed with comparison to the vector control strains of BL21-pET45b(+)/BL21(DE3)-pET45b(+) and expression control strain provided by supplier. Results indicated the r- SDUG was only expressed inE. colistrain BL21(DE3) but not in BL21 (data not shown) which was further confirmed by western blot analysis (data not shown) and BL21(DE3)-pET45b(+)-SDUG strain was used for the further studies. Expression control strain revealed a prominent band on SDS-PAGE for induced sample, indicating experimental protocols/conditions used are optimized. Further, these analyses also indicated recombinant protein is mainly concentrated into insoluble fraction by forming inclusion bodies and leaving a little protein in the soluble fraction. Therefore, attempts were made to optimize the expression conditions of r- SDUG in pET system with regard to IPTG concentration and temperature of incubation. Expression was carried out at the temperatures of 25, 28, 30 and 33 ℃and induction IPTG concentrations of 0.4 mM, 0.6 mM and 1 mM. Samples were taken in 1 h, 3 h, 5 h and 7 h and analyzed by SDS-PAGE. In these analyses, it was not revealed any satisfactory level of r-SDUG expression in soluble fractions compared to insoluble fractions (results are not shown) indicating r-SDUG is aggregate-prone and less hydrophilic and more hydrophobic protein.
E. coliis the most commonly used prokaryotes as a host for the expression of recombinant proteins. However, the production of foreign proteins inE. colioften aggregate to form insoluble inclusion bodies because of their inability to form native or correct tertiary structures due to anomalies in protein folding[12]. Co-expression of chaperones with the protein of interest is a good solution to overcome these problems and chaperone increases recovery of expressed proteins in the soluble fraction by helping correct protein folding. The most abundant and physiologically important chaperones inE. coliinclude DnaK, DnaJ, GrpE, GroEL, GroES[13,14] and additional factor of trigger factor[15]. These chaperones play distinct but cooperative role in protein folding in-vivo[16-18]. In this study, plasmids pG-KJE8, pGro 7, pKJE7, pG-Tf2 and pTf16 that express chaperone proteins of dnaK-dnaJ-grpE-groES-gro-E, groES-groEL, dnaK-dnaJ-grpE, groES-groEL-tig, and tig[9], respectively, involved in protein folding ofE. coliwere co-expressed with SDUG using optimized conditions of IPTG concentration (0.6 mM), incubation temperature (28 ℃) and duration of incubation (3 h) to study the optimal concentrations of L-arabinose and tetracycline that drive the expression of chaperones to give rise the highest soluble expression of r-SDUG. This approach revealed a significant improvement of soluble expression of r-SDUG, despite the larger portion of proteins still to be remained in the insoluble fraction. Of the five different chaperon plasmids employed, the accumulation of higher amount of r-SDUG to both the soluble and insoluble fractions in every concentration of L-arabinose was seen, when SDUG co-expressed together with pGro7 chaperone plasmid. However, the accumulation of r-SDUG into soluble and insoluble fraction seems to depend on the concentration of L-arabinose in the culture medium and the highest amount of recombinant protein in soluble fractions compared to the same in the insoluble fractions was seen in the presence of 2 mg/mL L-arabinose. On the contrary, the same chaperone plasmid produced considerably higher amount of r-SDUG in the insoluble fraction in the presence of 0.5 mg/mL L-arabinose and further, the amount of r-SDUG in both fractions seems to significantly reduce in the presence of 4 mg/mL L-arabinose. Furthermore, the r-SDUG in the insoluble fraction was generally higher in all types of chaperones at low concentration of L-arabinose and/or tetracycline indicating accumulation of protein of interest either to the soluble and insoluble fractions depends on the concentration of L-arabinose and/or tetracycline used in the culture medium.
Ammonium sulfate precipitation is a method used to purify proteins by altering their solubility known as salting out. At sufficiently high ionic strength, the protein will be almost completely precipitated from the solution. Since saltingout is a very useful procedure to assist in the purification of a given protein. However, in this method was not useful in purification of r-SDUG as precipitation was seen at all level of ammonium sulfate saturation. Therefore, Ni affinity chromatography was used as r-SDUG is Polyhistidinetagged at the N-terminal end of the protein. Polyhistidinetagged protein production in bacterial system allows the purification of proteins using Ni-NTA affinity column in a fast and easy manner. Further, histidine tags can be cleaved with protease cleavage and removed by affinity chromatography. But polyhistidine residues are always unnecessary to remove from the expression protein asprevious experiments showed that His-tag does not affect the bioactive activity of the recombinant protein[5,16] or the structure and function of the recombinant protein[19]. Polyhistidine-tagged r-SDUG was successfully purified using Ni-NTA column chromatography in both denaturing and native conditions and they were confirmed by western blotting with anti-His tag antibody.
Recombinant protein purified from insoluble fraction in denaturing conditions in this study will be used as an antigen to produce polyclonal antibodies against r-SDUG to detect tissue localization of this protein inS. digitataby means of immunohistochemical staining and the protein purified from soluble fraction in native conditions following chaperone mediated co-expression will be used for the determination of 3D-structural properties of SDUG. Finally, this study indicates for the first time thatS. digitataproteins can be expressed in bacterial system while demonstrating enhancement of soluble expression of aggregate-prone less hydrophilic and more hydrophobic r-SDUG using plasmid mediated chaperon co-expression.
Conflict of interest statement
The authors declare no conflict of interest.
Acknowledgements
This work was supported by a grant (SIDA/2006/BT/04) awarded by National Science Foundation of Sri Lanka.
[1] Tung KC, Lai CH, Ooi HK, Yang CH, Wang JS. Cerebrospinal setariosis with Setaria marshalli and Setaria digitata infection in cattle. J Vet Med Sci 2003; 657: 977-983.
[2] Dassanayake RS, Rodrigo WWP, Karunanayake EH, Weerasena OVDSJ, Chandrasekharan NV. A putative nuclear growth factorlike globular nematode specific protein. Bioinformation 2009; 3: 370-374.
[3] Baneyx F. Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol 1999; 10: 411-421.
[4] Betiku E. Molecular chaperones involved in heterologous protein folding in Escherichia coli. Biotechnol Mol Biol Rev 2006; 1: 66-75.
[5] Liu QH, Huang J, Han WJ, Liang Y, Lu CL, Wang QY. Expression, purification and characterization of WSSV-VP37 in Pichia pastoris. Aquaculture 2006; 258: 55-62.
[6] Martinez-Alonso M, Garcia-Fruitos E, Ferrer-Miralles N, Rinas U, Villaverde A. Side effects of chaperone gene co-expression in recombinant protein production. Microb Cell Fact 2010; 9: 64.
[7] Novy R, Yaeger V, Held D, Mierendorf R. Innovations 2002; 12: 2-6.
[8] Sambrook J, Russell DW. Molecular cloning-a laboratory manual. 3rd ed. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001.
[9] Chaperone Plasmid Set, Catalog number 3340, TaKaRa Bio Inc.
[10] Green AA, Hughes WL. Methods in enzymology. New York: Academic Press Inc; 1955, p.76.
[11] pET system manual. 11th edition. Novagen; 2005.
[12] Nishihara K, Kanemori M, Kitagawa M, Yanagi H, Yura T. Chaperone coexpression plasmids: differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli. Appl Environ Microbiol 1998; 64: 1694-1699.
[13] Georgopoulos C, Liberek K, Zylicz M, Ang D. Properties of the heat shock proteins of Escherichia coli and the autoregulation of the heat shock response. In: Morimoto RI, Tissieres A, Georgopoulos C, (eds.). The biology of heat shock proteins and molecular chaperones. New York: Cold Spring Harbor Laboratory Press, Cold Spring Harbor; 1994, p. 209-249.
[14] Gross CA. Function and regulation of the heat shock proteins. In: Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, et al. (eds.), Escherichia coli and Salmonella: cellular and molecular biology. Washington, D.C: AMS Press; 1996, p. 1382-1399.
[15] Nishihara K, Kanemori M, Yanagi H, Yura T. Overexpression of trigger factor prevents aggregation of recombinant proteins in Escherichia coli. Appl Environ Microbiol 2000; 66: 884-889.
[16] Gragerov A, Nudler E, Komissarova N, Gaitanaris GA, Gottesman ME, Nikiforov V. Cooperation of GroEL/GroES and DnaK/DnaJ heat shock proteins in preventing protein misfolding in Escherichia coli. Proc Natl Acad Sci USA 1992; 89: 10341-10344.
[17] Hartl FU. Molecular chaperones in cellular protein folding. Nature 1996; 381: 571-580.
[18] Kusukawa N, Yura T. Heat shock protein GroE of Escherichia coli: key protective roles against thermal stress. Genes Dev 1988; 2: 874-882.
[19] Ramos CRR, Abreu PAE, Nascimento ALTO, Ho PL. A highcopy T7 Escherichia coli expressionvector for the production of recombinant proteins with a minimal N-terminal His-tagged fusion peptide. Braz J Med Biol Res 2004; 37: 1103-1109.
*Corresponding author: Prof. R. S. Dassanayake. Department of Chemistry, Faculty of Science, University of Colombo, 90, Cumaratunga Munidasa Mawatha, Colombo 03, Sri Lanka.
Tel/Fax: 94-11-2503367
E-mail: rsdassanayake@chem.cmb.ac.lk
Foundation project: This work was supported by a grant (SIDA/2006/BT/04) awarded by National Science Foundation of Sri Lanka.
Uncharacterized gene
Chaperon
Co-expression
 Asian Pacific Journal of Tropical Medicine2014年2期
Asian Pacific Journal of Tropical Medicine2014年2期
- Asian Pacific Journal of Tropical Medicine的其它文章
- Bond strength analysis of the bone cement- stem interface of hip arthroplasties
- Hepatic effect of NAC on sevear acute pancteatise of rats
- Comparative analysis of different cyclosporine A doses on protection after myocardial ischemia/reperfusion injury in rat
- Comparison on serum biomarkers for anovulatory and ovulatory dysfunctional uterine bleeding in Lizu females
- Preparation of novel biodegradable pHEMA hydrogel for a tissue engineering scaffold by microwave-assisted polymerization
- Mathematical modeling for selecting center locations for medical and health supplies reserve in Hainan Province