
近8年我国药品监督管理部门发文修订药品说明书情况及趋势
2014-03-09 03:33夏东胜国家食品药品监督管理总局药品评价中心北京100045
中国药房 2014年41期
夏东胜(国家食品药品监督管理总局药品评价中心,北京 100045)
药品说明书是指导医师开具处方和患者用药的具有法律效力的文本,应当包含药品安全性、有效性的重要科学数据、结论和信息,以指导临床安全、合理用药[1]。药品说明书的内容是在上市前研究的基础上,由生产企业提出、药品监督管理部门(简称“药监部门”)核准的,其相关信息并非一成不变。随着药品在临床被广泛使用、药品不良反应(ADR)监测的开展及进一步深入研究,必然会积累更多安全性、有效性等方面的信息,需及时对说明书进行补充和完善。截至2013年12月,我国上市后药品有国产化学药批准文号106 519 个、中成药批准文号59 718 个、进口药品批准文号4 469 个[2]。我国药品说明书主要存在缺项、信息不完善等问题,国产药品说明书中“缺项”比例明显高于进口与合资药品,中成药说明书中“缺项”高于化学药[3]。因此,修订药品说明书成为完善药品信息、防控药品风险、促进合理用药的重要措施。我国于2006年6月1日起开始实施《药品说明书和标签管理规定》(简称“24号令”),促进了药品说明书对临床安全用药的指导性,提升了对公众用药安全的保障。本文就“24号令”实施后至2013年底我国药监部门发文要求企业修订药品说明书的情况进行分析,并探讨其趋势,以期为生产企业及相关部门做好药品说明书管理提供借鉴。
1 我国药品说明书制定与修订的相关法规依据
1.1 药品说明书的制定
《药品注册管理办法》[4]第143 条规定,药品说明书和标签由申请人提出,原国家食品药品监督管理局(SFDA)药品审评中心根据申报资料对其中除企业信息外的内容进行审核,在批准生产时由原SFDA 予以核准。申请人应当对药品说明书和标签的科学性、规范性与准确性负责。第145 条规定,申请人应当按照原SFDA规定的格式和要求、根据核准的内容印制说明书和标签。“24号令”第11条也指出,药品说明书应当列出全部活性成分或组方中的全部中药药味。注射剂和非处方药(OTC)还应当列出所用的全部辅料名称。药品处方中含有可能引起严重ADR的成分或辅料的,应予以说明;药品说明书的具体格式、内容和书写要求由原SFDA制定并发布。
“24号令”实施后,原SFDA相继发布了《关于印发化学药品和生物制品说明书规范细则的通知》[5]、《关于印发中药、天然药物处方药(Rx)说明书格式内容书写要求及撰写指导原则的通知》[6]和《关于印发非处方药说明书规范细则的通知》[7],为药品说明书的制定与修订提供了具体的技术规范和指导意见。
1.2 药品说明书的修订
《药品注册管理办法》第144条规定,申请人应当跟踪药品上市后的安全性和有效性情况,及时提出修改药品说明书的补充申请。“24号令”第12条规定,生产企业应当主动跟踪药品上市后的安全性、有效性情况,需要对药品说明书进行修订的,应当及时提出申请。根据ADR监测、药品再评价结果等信息,原SFDA也可要求生产企业修订药品说明书。
1.3 相关法律责任
“24号令”第14条指出,药品说明书应当充分包含ADR信息,详细注明ADR。生产企业未根据药品上市后的安全性、有效性情况及时修改说明书或未将ADR 在说明书中充分说明的,由此引起的不良后果由该生产企业承担。
《ADR 报告和监测管理办法》[8]第45 条指出,生产企业应对收集到的ADR报告和监测资料进行分析、评价,并主动开展药品安全性研究。生产企业对已确认发生严重ADR 的药品,应当通过各种有效途径将ADR、合理用药信息及时告知医务人员、患者和公众;采取修改标签和药品说明书,暂停生产、销售、使用和召回等措施,以减少和防止ADR的重复发生。还应当将药品安全性信息及采取的措施报所在地省级药品监督管理部门和原SFDA。第49条规定,原SFDA根据药品分析评价结果,可要求企业开展药品安全性、有效性相关研究。必要时,应当采取责令修改药品说明书,暂停生产、销售、使用和召回药品等措施。“24 号令”第13 条规定,药品说明书获准修改后,生产企业应当将修改的内容立即通知相关药品经营企业、使用单位及其他部门,并按要求及时使用修改后的说明书和标签。
2 近8年药监部门要求生产企业修订药品说明书发文情况
2.1 基本情况
截至2013年底,药监部门针对药品说明书修订发文合计85 个,近两年发文总数明显上升,覆盖了由单一药品、药品制剂到某一特定类别的药品,具体见表1(“*”表示包含针对具体药品和类别2 种形式,具体统计合计数时按1 个统计)。其中,涉及中药与化学药、Rx 与OTC 及说明书修订形式的发文分类情况见表2(“*”表示包括提供OTC 说明书范本与Rx 说明书修订要求2 种形式,具体统计合计数时按1 个统计)。发文涉及的药品剂型大部分为口服制剂,其次为注射剂,也有少数外用制剂,个别为直肠给药、阴道黏膜给药、局部口腔用药等,具体见表3(“*”表示同时包括中药、化学药内容)。
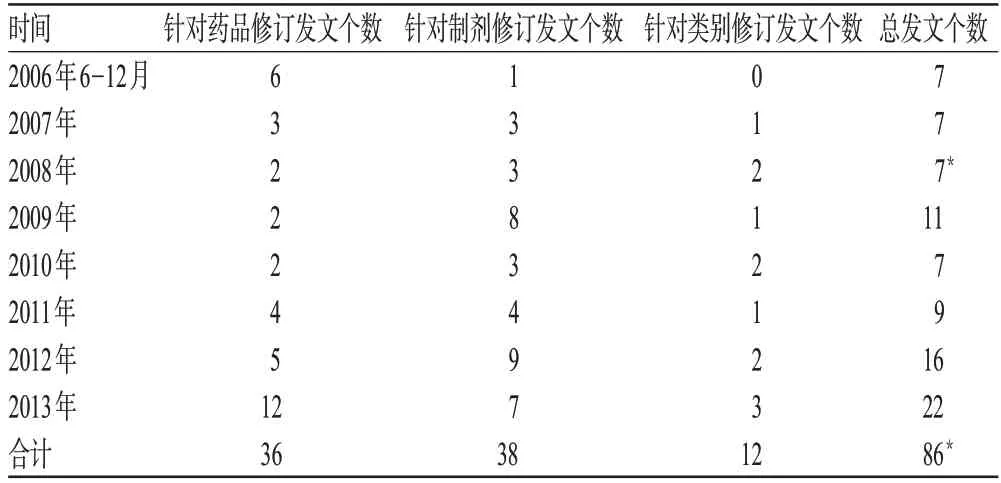
表1 药监部门要求企业修订药品说明书各年度发文情况Tab 1 Official documents that the drug regulatory authority requires manufacturers to revise drug package inserts

表2 发文分类情况Tab 2 The classification of documents
2.2 修订事项
2.2.1 修订要求中涉及项目情况。按照发文中说明书修订项目统计,52个提供修订要求(内容/说明)的发文中,明确涉及安全性项目修订的内容有125个,一般项目18个(见表4)。在注意事项中,也包含对特殊人群用药安全的提示,在涉及的一般项目中也有部分是基于安全性考虑所进行的修订。可见,安全性项目的修订为药监部门发文修订说明书的主要内容。
2.2.2 涉及多次发文的药品/制剂及其修订事项。某些药品因安全性原因,药监部门曾多次发文要求进行说明书修订,如:(1)2008年6月11日、2011年5月15日分别针对尼美舒利口服制剂说明书进行修订;2011年7月21 日针对尼美舒利缓释制剂说明书进行修订;(2)2008年7月7日、2008年7月21日分别针对非甾体抗炎药Rx 和部分OTC 品种说明书进行修订;(3)2008年11月19日、2012年5月18日分别针对拉莫三嗪片和盐酸拉莫三嗪口服制剂说明书进行修订;(4)2009年4月2 日、2012年4月24 日分别针对盐酸吡格列酮制剂说明书进行修订;(5)2010年9月1日、2012年4月24日分别针对奥利司他制剂说明书进行修订;(6)2010年11月24 日、2012年12月14 日分别针对左炔诺孕酮片非处方药说明书进行修订。由此可见,说明书多次修订的原因主要是药品安全信息的不断更新,也反映了药监部门对公众用药安全的重视。

表3 发文涉及品种情况Tab 3 Involved types in documents

表4 修订要求中涉及项目情况Tab 4 The items involved in the revision requirements
2.2.3 并用的其他监管措施。(1)修订说明书后停止生产销售使用的药品制剂1 个(盐酸丁咯地尔制剂);(2)修订说明书涉及ADR通报的品种23个(其中化学药15个、中药8个);(3)修订说明书同时转为Rx的发文2个(即将含麻黄碱类复方制剂6个药品和养血生发胶囊等5 个含何首乌的药品转为Rx 管理)。(4)将甲类OTC 转为乙类OTC 同时修订说明书发文1 个(即将归脾颗粒等7个品种由甲类OTC转换为乙类OTC)。
2.3 药品说明书修订的原因
2.3.1 基于对药品安全性监测与评价所获得的药品风险信息所进行的修订。根据原SFDA监测评价结果,为控制药品使用风险、保障公众用药安全,修订药品说明书,增加有关提示信息;或根据ADR评估结果,为控制药品使用风险、保证用药安全有效等原因,决定对说明书进行修订。此类信息包括国内外收集的药品安全性信息。如罗格列酮及其复方制剂、非甾体抗炎药说明书修订等主要是源于国外安全信息的修订;养血生发胶囊等6 个含何首乌中药口服制剂说明书修订是源于国内安全性信息的修订。
2.3.2 基于药品标准变更情况所进行的修订。某些药品说明书修订文件中提到:“为保证公众用药安全,根据《药品管理法》及其实施条例、《Rx与OTC分类管理办法(试行)》的有关规定以及药品标准变更等情况,原SFDA 对部分OTC 说明书范本进行修订”。如原SFDA对《中国药典》2010年版或其他标准收载的OTC,依据其标准变更情况对说明书范本进行的修订。
2.3.3 基于对说明书的规范与完善、统一表述所进行的修订。如硫普罗宁最初上市的说明书内容较简单,适应证不规范,ADR和注意事项等安全性信息较少;已上市的注射用盐酸头孢替安、核黄素磷酸钠注射液和注射用核黄素磷酸钠的说明书有多个版本,其中的适应证、用法用量、药动学等内容差别较大,为保证患者用药安全对说明书进行规范等。
2.3.4 基于说明书相关法规或规范改变所进行的修订。如“24 号令”及《OTC 说明书规范细则》发布后,为进一步规范OTC 说明书和标签的管理,原SFDA 组织的对已公布的OTC品种说明书范本按照相关规范进行的修订。
2.3.5 基于对药品分类管理状态的改变所进行的修订。如根据《关于印发药品类易制毒化学品专项整治行动实施方案的通知》和《关于加强含麻黄碱类复方制剂管理有关事宜的通知》要求,麻黄碱含量超过30 mg的复方制剂从OTC转为Rx管理。依据Rx的管理要求,原SFDA 对相关6个Rx说明书进行核准;为保障公众用药安全,将养血生发胶囊等5 个含何首乌的OTC 转为Rx 管理,并对其说明书按照Rx 要求进行了修订。此外,还将归脾颗粒等7 个品种由甲类OTC 转换为乙类OTC,并对其OTC说明书范本进行修订。
2.3.6 基于勘误或更正信息所进行的修订。如《关于修改磷霉素钙片说明书用法用量项的通知》中,因说明书用法用量有误进行的修订;《关于修订消肿止痛酊等品种非处方药说明书范本的通知》中,对人参补丸(胶囊)、氨酚伪麻那敏口服溶液用法用量及注意事项内容的修订等。
2.4 对企业修订说明书程序和时限的要求
目前,药监部门要求生产企业对药品说明书修订的文件中,对说明书修订的具体程序和时限也提出了具体要求。对因安全性方面原因提出修订的具体规定一般为:“在某年某月某日前,依据《药品注册管理办法》等有关规定提出修订说明书的补充申请报备案”“补充申请备案之日起生产的药品,不得继续使用原药品说明书”“应当将说明书修订的内容及时通知相关医疗机构、药品经营企业等单位,并在补充申请备案后6个月内对已出厂的药品说明书予以更换”[9]。其中,对修订说明书补充申请报备案的时间规定一般是1~3个月。
值得一提的是,原SFDA既往发文中并没有具体时间的规定,而是要求生产企业“尽快修订说明书及标签的相关内容,按照有关规定进行备案”“并尽快对已出厂的药品说明书予以更换”。生产企业应当将修订的内容及时通知相关医疗机构、药品经营企业等单位[10]等。可见,有具体时间限定的管理模式更具可操作性,有利于发挥有效监管的作用。
3 我国药品说明书修订的趋势
药品说明书的科学规范是保障消费者安全用药的重要手段,是防止滥用、误用的重要屏障。消费者有权利做出如何从药品得到最大疗效和防止损害的知情决定。据调查显示,绝大部分公众会在用药前阅读药品说明书,掌握一些所服药品的基本信息[11]。部分患者由于用药前阅读说明书后对药品产生怀疑,还可能提出退药要求。我国十分重视对药品说明书的管理,近年来发布实施了一系列相关法规及文件,对药品说明书的法制化、科学化、规范化起到了重要作用。随着我国公众对药品说明书信息需求的不断提升及全球药监部门对药品说明书安全性信息的日益关注,我国未来药品说明书信息修订的趋势还将不断加强。
3.1 药品说明书中安全性信息需进一步完善
由本文统计的“24号令”实施后我国药监部门发文修订药品说明书的情况看,近年来我国已对药品说明书中安全性信息给予了重视并努力不断使其完善。对于我国特色的中药而言,主要是基于我国药品上市后使用、监测与研究的基础上进行的修订,体现了我国药品说明书管理的特色,近年来已针对某些中药注射剂、含毒性药材药品、现代研究发现有安全性问题的某类药材及其制剂、某些中西复方制剂等修订了说明书;对于化学药而言,主要是结合国外监管的安全性信息与我国的监测数据对说明书进行修订。近年来,对某些发现有较大安全风险的化学药甚至还多次发文修订说明书的安全性信息。但以上措施,相对于我国药品说明书的整体水平而言,还远远不够,药品说明书安全性信息的修订与完善还需向常态化和精细化发展,任重而道远。
3.2 药品说明书的规范性需不断加强
我国仿制药居多,相同Rx存在不同的规格或剂型,由于药品获得批准的时间、生产企业不同,所进行的研究及结果差异,致使我国不同企业之间同种成分的药品说明书的内容不尽相同,不仅给医师用药带来不便甚至困惑,也使患者在药品使用过程中遇到一些实际困难[12]。
此外,不同企业版本的说明书中重要项目及信息的缺失,也可能对患者用药造成影响。目前,我国药监部门已因上市后不同版本说明书差异问题多次发文,对说明书重要内容进行统一和规范,但鉴于我国上市后药品的特点,对药品说明书的规范还需做更多的努力。
3.3 药品说明书修订的范围更加广泛
由近8年我国药监部门发文修订说明书的情况看,涉及中药与化学药、Rx与OTC,及多种剂型与用药途径药物等。药品说明书修订发文中既包含单个具体药品或同方制剂药品,也包含对某一特定类别的药品,如含毒性中药饮片中成药品种、非甾体抗炎药等。鉴于我国上市后研究及ADR 监测的发展,今后由单一药品评价向类别药品评价的展开,在数据信息基础上采取相应的修订说明书的宏观措施,将致使说明书修订的范围更加广泛。
3.4 药品说明书修订的监管力度需更加有效合理
由本文统计结果可见,我国对企业修订药品说明书及标签的申请备案、对已出厂药品说明书的更换时限经历了从无到有的探索过程。药监部门对企业药品说明书修订的具体程序正由粗放式向定向式转变,使其更具操作性,监管更加有效,但有关规定的时限是否合理,还需进一步探索。此外,依据药品的安全风险,药品说明书修订还可继续伴随药品其他监管措施的使用,如停止生产与销售、ADR信息通报、转为Rx管理等,也反映了药品安全监管力度的加强。
3.5 药品说明书修订中的法律责任主体应更加凸显
《药品注册管理办法》、“24号令”及《ADR报告和监测管理办法》均对说明书修订中生产企业的责任予以了明确。因此,生产企业是药品安全的第一责任人,是修订药品说明书的主体,未来需真正体现与强化在药品说明书修订中的主体责任。此外,国家药监部门依据获得的药品信息也可责令企业修订药品说明书。
4 结语
总之,在今后药品说明书修订过程中,生产企业的主体责任需进一步强化,应变目前普遍存在的被动修订为主动负责任的修订;在加强药品监测与研究基础上,使说明书内容更趋完善,增强药品使用的可指导性,有效控制药品安全风险。药监部门应构建有效信息沟通的平台,对于已获知的重要安全风险等共性问题采取相应措施,可责令企业进行说明书修订。此外,药监部门的另一重要职责是在说明书有效监管方面做出更多努力。相信随着企业主体责任的强化,未来我国药品说明书修订的力度必将受到各方重视与不断加强,更好地发挥保障公众安全用药的作用。
[1]国家食品药品监督管理局.药品说明书和标签管理规定[S].2006-03-15.
[2]国家食品药品监督管理总局.数据查询[EB/OL].(2013-12-31)[2013-12-31].http://appl.sfda.gov.cn/datasearch/face3/base.jsp.
[3]路俊华,祝培友,孙兆勇,等.药品说明书安全事项调查分析[J].中国医药导报,2010,7(16):190.
[4]国家食品药品监督管理局.药品注册管理办法[S].2007-07-10.
[5]国家食品药品监督管理局.关于印发化学药品和生物制品说明书规范细则的通知[S].2006-05-10.
[6]国家食品药品监督管理局.关于印发中药、天然药物处方药说明书格式内容书写要求及撰写指导原则的通知[S].2006-06-22.
[7]国家食品药品监督管理局.关于印发非处方药说明书规范细则的通知[S].2006-10-20.
[8]国家食品药品监督管理局.药品不良反应报告和监测管理办法[S].2011-05-04.
[9]国家食品药品监督管理总局.国家食品药品监督管理总局办公厅关于修订灯盏花素注射液与注射用灯盏花素说明书的通知[S].2013-12-02.
[10]国家食品药品监督管理局.国家食品药品监督管理总局关于修订雷公藤中成药说明书的通知[S].2012-10-18.
[11]冯变玲,杨世民,叶竹松.公众对药品不良反应认知度的调查报告[J].医药导报,2010,29(12):1 667.
[12]翟所迪,杨波.从安全性角度对药品说明书异同的调查分析:一[J].中国医院用药评价与分析,2008,8(1):76.
猜你喜欢
中学生天地(A版)(2022年6期)2022-07-14
好日子(2021年8期)2021-11-04
今日农业(2020年18期)2020-12-14
戏曲研究(2020年1期)2020-09-21
今日农业(2020年14期)2020-08-14
作文成功之路·小学版(2020年3期)2020-04-21
中成药(2017年4期)2017-05-17
今日农药(2017年2期)2017-03-24
