
碳酸钙超细研磨用分散剂的制备与应用
2014-01-12 10:47胡惠仁何秋实
中国造纸 2014年4期
张 雪 胡惠仁 何秋实
(天津科技大学天津市制浆造纸重点实验室,天津,300457)
近年来,造纸涂布技术的发展,对涂布颜料提出了越来越高的要求[1-2]。碳酸钙来源广泛、成本低、白度高、吸墨性好,在造纸涂布颜料领域得到了越来越广泛的应用,特别是天然研磨碳酸钙 (GCC)用量剧增,主要与以下4方面原因相关:①由于超细研磨技术的发展,使天然碳酸钙粒子细度使纸张涂层光泽度等于或超过沉淀碳酸钙 (PCC)。②我国天然方解石资源储量丰富,原料来源便捷,制造成本远低于高岭土等其他涂布颜料。③天然碳酸钙对涂布胶黏剂需要量少,可降低胶黏剂费用。④碳酸钙有良好的流变性,涂料可以做到高固含量,节省能耗[3-8]。
随着涂布加工技术的发展,涂布级超细GCC的粒度从10年前的小于2 μm达90%的要求,发展到现在市场上经常见到的小于2 μm达98%,甚至99%,而且对超细GCC的粒径分布要求也越来越高。这就对研磨专用设备和研磨专用分散剂提出了越来越高的要求。在研磨阶段,研磨分散剂分为前段分散剂和后段分散剂。后段分散剂由于市场需求、附加值高等原因,引起了广大造纸工作者越来越浓厚的兴趣,特别是丙烯酸类分散剂[9-10]。
分散剂是靠吸附在无机颜料表面,产生空间位阻[11-12]和静电斥力[13],从而使颜料粒子能以悬浮态稳定地存在,才能得到高固含量、低黏度的GCC浆料,研磨效率也得以提高,并能节约研磨能耗。
本研究以丙烯酸 (AA)、2-丙烯酰胺-2甲基丙磺酸 (AMPS)为聚合单体,过硫酸铵为引发剂,次亚磷酸钠为链转移剂,采用水溶液自由基聚合方法,制备GCC超细研磨用后段分散剂二元聚合物P(AA-AMPS);通过单因素实验优化出最佳的合成条件,并将合成的后段分散剂与市售产品进行对比。
1 实验
1.1 主要原料与仪器
AMPS:工业级;AA、过硫酸铵、次亚磷酸钠、氢氧化钠:均为分析纯;325目GCC:天津燕东公司;前段分散剂 (M):永港伟方 (北京)科技股份有限公司;后段分散剂样品 (市售):日本产后段分散剂LX-7,国内某厂后段分散剂MN-9。
DZKW-1型电热恒温水浴锅:北京永光明医疗仪器厂;BT/1011型蠕动泵:保定雷弗流体科技有限公司;S211-90型恒速电动搅拌器:上海豫康科教仪器设备有限公司;篮式研磨机:上海索维机电公司。Sower实验室分散机:上海索维机电公司。
DV-II+型旋转黏度计:Brookfield(博勒飞)公司;LS13 320激光衍射粒度分析仪:美国贝克曼库尔特公司。Vector22型傅里叶变换红外光谱仪:德国布鲁克光谱仪公司;AVANCE型核磁共振波谱仪:布鲁克拜厄斯宾有限公司;凝胶渗透色谱-十八角度激光散射联用系统:美国怀雅特技术公司。
1.2 P(AA-AMPS)分散剂的合成
将一定量的水、氢氧化钠加入到带有温度计的四口烧瓶中,置于预先设定好温度的水浴锅中,并与搅拌器连接。将次亚磷酸钠及过硫酸铵分别配置成一定浓度的溶液备用;称取一定量的水、AA及AMPS于容器中,待固体溶解后备用。待水浴锅升至合适温度后,将过硫酸铵溶液、次亚磷酸钠溶液及混合单体溶液用蠕动泵滴加,待溶液滴加完毕后,使溶液在一定温度下保温一段时间。即可得到P(AA-AMPS),产物为淡黄色透明液体。
1.3 P(AA-AMPS)性能分析
固含量:样品于105℃下烘干至恒质量,计算固含量。
特性黏度的测定:用乌氏黏度计 (Ubbelohde viscometer)按照国家标准 GB/T 1200.5.1—1989 及GB/T 10533—2000,采用稀释法,用101 g/L的硫氰酸钠溶液作为溶剂测定聚合物溶液的乌氏黏度。
电荷密度的测定:准确量取1mL一定浓度的P(AA-AMPS)溶液置于MÜTEK PCD 03 pH-S圆筒测量室内,加水至测量室充满,待读数稳定后,用标准阳离子液PS-Na滴定至电位为零,记录标准阳离子液的用量,电荷密度按公式 (1)计算。
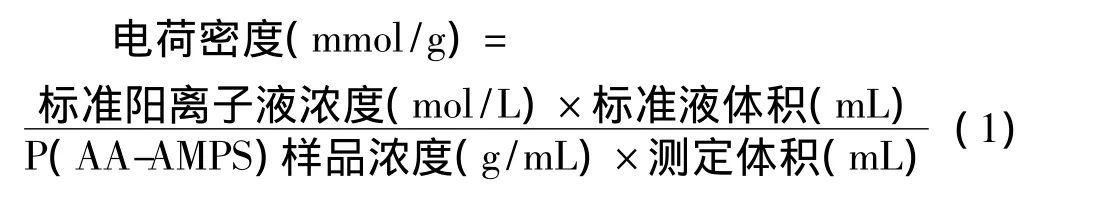
傅里叶红外光谱分析 (FT-IR):将合成的P(AA-AMPS)分散剂用乙醇进行沉淀,抽滤后的粗产物,再用乙醇洗涤数次,洗去未反应的单体,静置后除去上层清液,将产物在40℃真空干燥24 h,得到纯化的聚合物,采用溴化钾压片法制样,然后进行FT-IR分析。
核磁共振波谱分析 (NMR):将合成的P(AAAMPS)分散剂用上述方法纯化后,将得到的固体研磨成细小粉末,以氘代水为溶剂,进行13C-NMR分析。
液相凝胶渗透色谱 (GPC):将产物按照上述方法纯化,进行GPC图谱分析。聚合产物的分子质量及分布的测定条件:以Waters 515 HPLC Pump单元泵为输液系统,以Shodex SB-806M为凝胶色谱柱,检测器为Wyatt-DAWN HELEOS-Ⅱ十八角度激光光散射仪和Wyatt-Optilab rex示差折光检测器联用,以0.1 mol/L硝酸钠溶液 (用1.0 mol/L氢氧化钠溶液调pH值至9.0)+0.02%叠氮化钠溶液为流动相。流动相经0.22 μm水系过滤膜低真空度过滤,测试柱温40℃,示差折光检测器温度 40℃,流速为0.5 mL/min,进样体积0.2 mL(满定量环进样)。
1.4 GCC浆液制备
以325目GCC为原料,先用前段分散剂M按相同研磨工艺研磨至粒径2 μm的含量占65%,再按一定的研磨工艺加入P(AA-AMPS)分散剂研磨一定时间,取样检测。
1.5 GCC浆液性能检测
GCC浆液的表观黏度测定:取适量的GCC浆液于量杯中,用旋转黏度计测定其表观黏度。即时黏度(T0):研磨结束后即时测定的悬浮液表观黏度,反应研磨结束时GCC浆液中颗粒间的电空间稳定性。静止黏度 (T24):静止24 h后测得的表观黏度,近似反应GCC浆液在长期运输或储存过程中的稳定性。动态黏度 (T'24):静止24 h后浆液经高速分散5 min后测得的表观黏度,反应静止存放的GCC浆液经过剧烈搅动后的黏度特性。回黏黏度 (T'24+1):静止24 h后的浆液经高速搅拌5 min再静止1 h测得的表观黏度,反应GCC浆液的流变性。
GCC浆液粒径及其粒径分布的测定:取一定量的GCC浆液,稀释至一定浓度,置于激光衍射粒度分析仪中,测定GCC的粒径及其粒径分布。
2 结果与讨论
2.1 单因素实验
以AA、AMPS为聚合单体,过硫酸铵为引发剂,次亚磷酸钠为链转移剂,进行水溶液自由基共聚合反应,制备GCC超细研磨用P(AA-AMPS)分散剂,对单体配比、引发剂用量、链转移剂用量、反应时间等反应参数进行单因素实验,考察了上述反应参数对聚合物电荷密度、特性黏度及其对GCC浆液研磨效果的影响。研磨实验中的空白实验指研磨后段继续加入前段分散剂的实验效果。
2.1.1 单体配比
AMPS是一种活性很高、具有大的侧基、分子质量较大、含—SO-3的聚合单体[14],共聚合反应过程中,AMPS用量不同,对聚合物的溶液性能及应用性能都有显著影响。单体配比对聚合物电荷密度和特性黏度的影响如图1所示。从图1可以看出,在单体总质量一定的情况下,随着AMPS用量的增加,P(AAAMPS)电荷密度迅速增加,当增加至一定值后,增速降低;特性黏度先下降后增加。这可能是因为,由于AMPS本身具有链转移剂的功能,随着AMPS用量的增加,自由基链增长过程受阻,已发生歧化终止,或与单体自由基发生耦合终止,降低了分散剂的相对分子质量,使分散剂的特性黏数降低,但随着AMPS用量的进一步增加,分散剂分子中含有的—SO-3基团增加,基团之间的静电斥力使得分子链构象伸展,增加了特性黏度。由于—SO-3基团是强阴离子型基团,随着AMPS用量的增加,分散剂中—SO-3基团含量增加,使得分散剂的电荷密度增加。
将不同单体配比制备的P(AA-AMPS)聚合物溶液应用于GCC浆液的研磨实验,其结果如图2所示。由图2可以看出,在单体总质量一定的情况下,随着AMPS用量的增加,静止黏度变化幅度最大,单体配比m(AMPS)∶m(AA)为0.25时最低;即时黏度在单体配比为0.25时为230 mPa·s,在单体配比为0.50时最大,为343.2 mPa·s;动态黏度在单体配比为0.5时最大;回黏黏度的变化较大,在单体配比为0.17与0.20时相近,在0.25时达到最低,之后增加的幅度变大,在0.50时达到2000 mPa·s。此外,还可以发现,只有单体配比为0.25时回黏黏度减少,其他条件下回黏黏度都比动态黏度大。这说明单体总质量中AMPS的含量对GCC浆液表观黏度变化有很大影响,特别是对回黏黏度。这可能与 P(AAAMPS)分子的电荷密度不同有关,太高的电荷密度反而不利于GCC浆液的后续稳定。综合分析实验结果,选择单体配比 m(AMPS)∶m(AA)=0.25。

图1 单体配比对P(AA-AMPS)电荷密度和特性黏度的影响
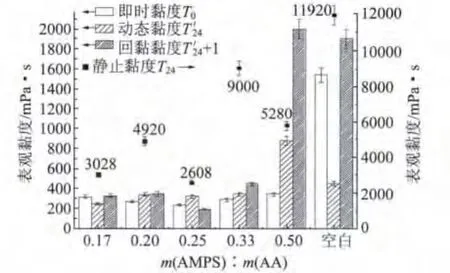
图2 单体配比对P(AA-AMPS)助磨性能的影响
2.1.2 引发剂用量
引发剂是结构上含有弱键的化合物,在热能或辐射能的作用下,会在弱键处断裂成自由基,从而引发聚合反应[15]。本实验采用过硫酸铵为引发剂,其用量对P(AA-AMPS)电荷密度和特性黏度的影响如图3所示。从图3可以看出,随着过硫酸铵用量的增加,P(AA-AMPS)的特性黏度先迅速下降,之后趋于稳定,电荷密度则先增加后下降。这主要是因为随着引发剂用量增加,聚合反应的活性中心的数量增加,当单体浓度一定时,使P(AA-AMPS)分子链变短,相对分子质量减少,从而P(AA-AMPS)的特性黏度下降,当引发剂用量增加至一定程度时,活性中心的数量不再增加,P(AA-AMPS)的特性黏度趋于稳定。

图3 引发剂用量对P(AA-AMPS)电荷密度和特性黏度的影响
将不同引发剂用量下制备的P(AA-AMPS)分散剂应用于GCC浆液研磨实验,结果如图4所示。由图4可以看出,随着引发剂用量的增加,静止黏度和回黏黏度都呈先减小后增大的趋势,静止黏度在引发剂用量5%(占单体总质量分数,下同)时最小,回黏黏度在引发剂用量4%时最小。即时黏度和动态黏度除在引发剂用量2%时较大外,其他条件下相差不大。综合分析实验结果,选择引发剂用量为4%。

图4 引发剂用量对P(AA-AMPS)助磨性能的影响
2.1.3 链转移剂用量
在合成P(AA-AMPS)的过程中,加入链转移剂控制链增长反应,使共聚物分子链长度适中。本实验选择次亚磷酸钠为链转移剂,其用量对聚合物电荷密度和特性黏度的影响如图5所示。从图5可以看出,随着链转移剂用量的增加,P(AA-AMPS)的特性黏度逐渐降低,电荷密度先增加后减小。这主要是因为,随着链转移剂用量的增加,体系中产生了大量的磷酸自由基,导致引发剂产生的活性自由基,首先与磷酸自由基进行反应而失去活性,使得引发聚合反应的活性自由基的数量减少,从而降低了P(AAAMPS)的特性黏度。此外,由于—SO-3基团活性高,聚合物分子链中磺酸基团的含量增加,磷酸基团和磺酸基团都是强阴离子基团,所以电荷密度逐渐增加,但当链转移剂用量增加至一定程度时,反应体系中链转移率很高,导致聚合物特性黏度迅速下降,电荷密度降低。
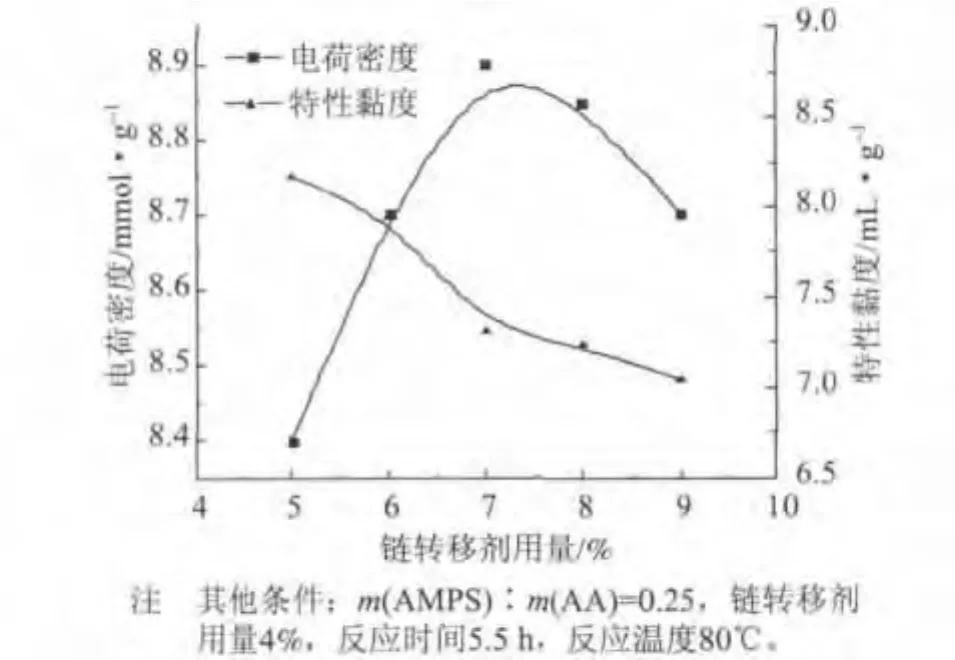
图5 链转移剂用量对P(AA-AMPS)电荷密度和特性黏度的影响
将不同链转移剂用量下制备的P(AA-AMPS)应用于GCC浆液研磨实验。实验结果如图6所示。由图6可以看出,随着链转移剂用量的增加,静止黏度变化幅度最大,在用量为7%(占单体总质量分数)时最低;即时黏度在用量为7%时最低为230 mPa·s,在其他用量时相差不大;动态黏度随着链转移剂用量的增加变化不大,但是回黏黏度浮动较大,在用量为7%的时候最小。这可能是由于在用量7%时,聚合物的电荷密度最高,特性黏度适中,能够提供GCC浆液研磨后段的电荷需要和包覆GCC颗粒所需要的分子质量。综合分析实验结果,选择链转移剂用量为7%。

图6 链转移剂用量对P(AA-AMPS)助磨性能的影响
2.1.4 反应时间
自由基聚合体系中,单体和聚合物同时存在,随着聚合反应的进行,单体含量逐渐降低,聚合物浓度增加,延长反应时间,可提高聚合产物的转化率[16]。反应时间对聚合物电荷密度和特性黏度的影响如图7所示。从图7可以看出,随着反应时间的增加,聚合物的特性黏度逐渐增加,电荷密度先增加后减小。这主要是因为反应时间增加,聚合物的转化率增加,特性黏度增加,分子链中磺酸基团含量增加,聚合物电荷密度增加,但随着反应时间增加至一定值,副反应增加,对电荷密度的增加产生负面影响,导致电荷密度下降。

图7 反应时间对P(AA-AMPS)电荷密度和特性黏度的影响
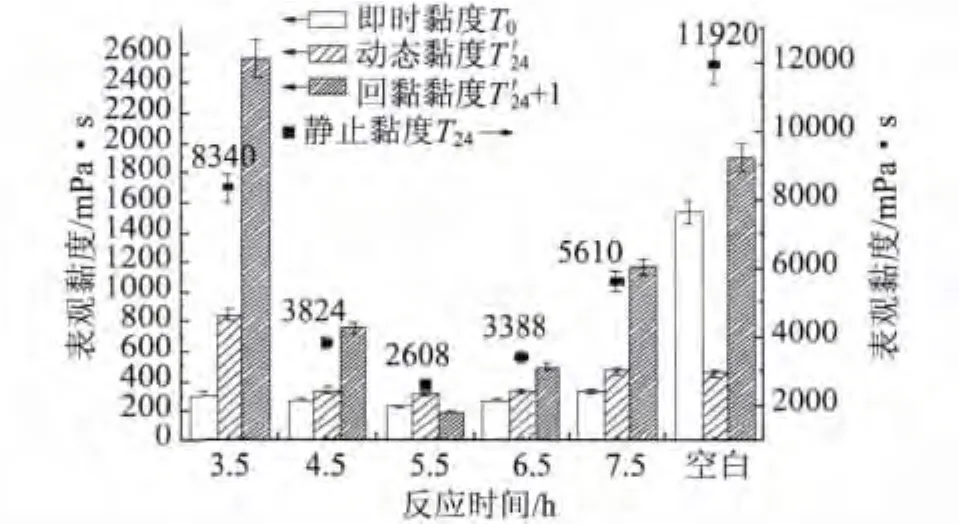
图8 反应时间对P(AA-AMPS)助磨性能的影响
将不同反应时间制备的P(AA-AMPS)应用于GCC浆液研磨实验。实验结果如图8所示。由图8可以看出,随着反应时间的增加,静止黏度变化幅度最大,呈先减小后增大的趋势,在聚合反应时间为5.5 h时最小。即时黏度随聚合反应时间的增加基本不变,都远小于空白样。动态黏度随聚合反应时间的增加基本呈先减小后增大的趋势,在聚合反应时间为4.5 h、5.5 h、6.5 h 时相差不大。回黏黏度随聚合反应时间的增加先减小后增加,在聚合反应时间为5.5 h时最低,在反应时间为3.5 h时回黏黏度最大。说明反应时间不足,对GCC浆液的稳定性有不利影响。综合分析实验结果,选择反应时间为5.5 h。
2.1.5 反应温度
研究表明[17],反应温度影响聚合反应速率和活化能,对产物的分子质量、共聚物分子结构都有很大的影响。对自由基聚合而言,引发剂分解是吸热反应,反应活化能高,约为125 kJ/mol,而初级自由基和单体加成反应打开π键是放热反应,活化能低,约2134 kJ/mol。因此自由基聚合具有慢引发、快增长的特点。温度太高,反应剧烈,不宜控制;温度太低,引发剂分解时间长,有时甚至不能引发反应。
反应温度对聚合产物特性黏度和电荷密度的影响如图9所示。从图9可以看出,随着反应温度的升高,产物的特性黏度先减小后增加,电荷密度先增加后减小。这可能是因为,随着反应温度的升高,引发剂受热分解的速率增加,活性自由基的生成速率随之增加,聚合速率加快,高分子链的增长受到抑制,引起特性黏度降低。反应温度继续增加,分子链容易发生交联,使聚合物特性黏度增加。随着反应温度的增加,—SO-3基团的反应活性增加,产物中—SO-3基团的含量增加,而反应温度增至一定值时,聚合反应发生严重的交联反应,故聚合物的电荷密度先增加后减小。

图9 反应温度对P(AA-AMPS)电荷密度和特性黏度的影响
将不同反应温度制备的P(AA-AMPS)应用于GCC浆液研磨实验。实验结果如图10所示。由图10可以看出,随着反应温度的增加,4种黏度指标基本都呈先减小后增大的趋势,静止黏度和回黏黏度变化幅度较大。在反应温度为80℃时各项黏度指标都达到了最低值。动态黏度随反应温度的增加变化最小。从图10还可以看出,反应温度为70℃、75℃、85℃和90℃时,回黏黏度都较大。这可能是由于聚合反应温度过低或过高,产物的特性黏度都较大,研磨后段容易在GCC颗粒间形成架桥,颗粒间发生团聚,且分散剂的电荷密度不够,不足以在颗粒间形成稳定的双电层,致使GCC浆液黏度增加。综合分析实验结果,选择反应温度为80℃。
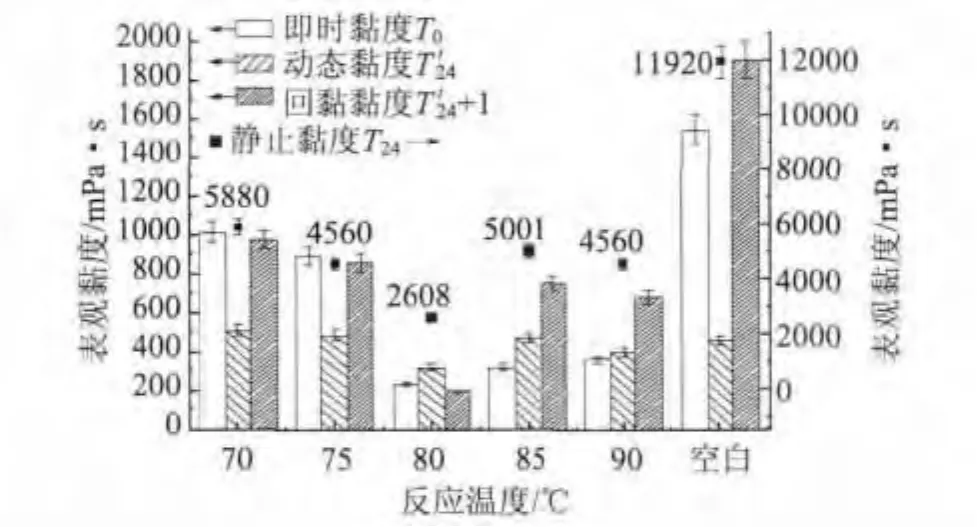
图10 反应温度对P(AA-AMPS)助磨性能的影响

图13 P(AA-AMPS)的GPC图谱

图11 P(AA-AMPS)分散剂的FT-IR图
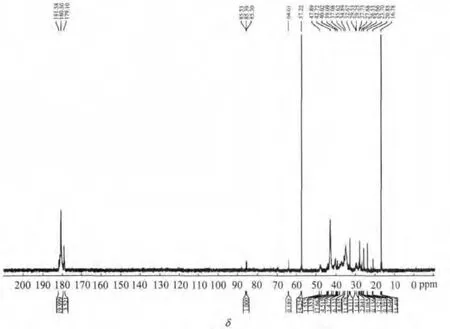
图12 P(AA-AMPS)分散剂的13C-NMR谱图
2.2 聚合物的性能表征
图11为P(AA-AMPS)的FT-IR图。由图11可以看出,=CC双键伸缩振动区(1690~1500 cm-1)无特征吸收峰,说明发生了自由基共聚反应。3432.67 cm-1处是酰胺基 (—CONH—)的伸缩振动吸收峰,2940.91 cm-1处是—CH3的伸缩振动吸收峰,1714.41 cm-1处是—=CO的伸缩振动吸收峰,1191.23 cm-1和 1039.44 cm-1处是—SO-3的伸缩振动吸收峰。FT-IR分析表明,实验所得产物确为AA和AMPS的二元共聚物。
图12所示为P(AA-AMPS)的13C-NMR谱图。由图12可以看出,σ为180.50是AA中—COO—羧基碳的化学位移;σ为35.86~37.08是AA中丙烯基上碳的化学位移;σ为32.09是AMPS中—CH3甲基碳的化学位移;σ为25.05是 AMPS中—CH2—亚甲基碳的化学位移;σ为40.02~42.89 是 AMPS 中—C+(CH3)2碳的化学位移。分析表明,实验所得产物确实为AA和AMPS二元共聚物。
图13为P(AA-AMPS)的GPC图谱。从图13可以看出,P(AA-AMPS)的GPC图谱只有一个较窄的峰,利用专门的GPC图谱分析软件计算聚合物的分子质量及多分散性系数如下:重均分子质量MW=6291,数均分子质量Mn=5424,Z均分子质量,Mz=7337,多分散性系数为1.160。实验表明,对于后段分散剂而言,较低的分子质量及较窄的分子质量分布有利于GCC浆液的后段研磨,有利于提高GCC浆液的分散稳定性及流动性能。因此,根据分子设计及自由基聚合机理,将聚合物的相对分子质量控制在5000左右。
将合成的P(AA-AMPS)经过纯化并真空干燥,置于带盖的坩埚中进行TG-DTG分析,测试条件如下:在N2氛围子下,升温速率为10℃/min,载气流速为50 mL/min,测定温度范围为室温至1000℃。实验结果如图14所示。从图14可以看出,P(AAAMPS)在238.2℃时开始分解,具有较好的热稳定性,能够适应GCC浆液研磨后段的高温环境,不会因为热分解而失效。
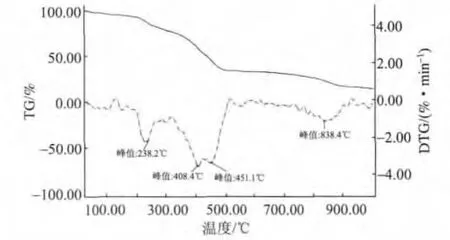
图14 P(AA-AMPS)的TG-DTG图
2.3 P(AA-AMPS)的应用
2.3.1 P(AA-AMPS)用量
GCC浆液后段研磨阶段,细小颗粒的含量增加,产生了许多新的断裂面,且研磨后期研磨槽内温度较高,前段分散剂已经不适合后段研磨,需要补充适量的后段分散剂来满足研磨后期的特殊条件。固定前段分散剂用量0.5%(对绝干GCC计,下同),实验了后段分散剂P(AA-AMPS)用量对GCC后段研磨的影响,结果如图15所示。由图15可以看出,随着P(AA-AMPS)用量的增加,GCC浆液的回黏黏度先减小后增加。这主要是因为,当P(AA-AMPS)用量低时,对于研磨新产生的断裂面,不能吸附足够的后段分散剂,以形成稳定的双电层。当P(AA-AMPS)用量过高时,新产生的断裂面吸附了足够的后段分散剂后,多余的分散剂会继续压缩双电层,使静电空间位阻作用减弱,从而对GCC浆液的稳定性带来负面影响。综合分析实验结果,选择后段分散剂P(AA-AMPS)最佳用量为1.2%。
2.3.2 添加方式

图15 P(AA-AMPS)用量对GCC浆液研磨效果的影响

图16 P(AA-AMPS)添加方式对GCC浆液研磨效果的影响
P(AA-AMPS)的添加方式对GCC浆液研磨效果的影响如图16所示。由图16可以看出,分段添加方式制备的GCC浆液各项黏度明显小于一次性添加的。这可能的原因是,由于分散剂一次性添加,对于研磨初期,分散剂是过量的;在研磨过程中,部分过量分散剂在高速剪切力和摩擦热的作用下分子结构被破坏;在后续研磨过程中,由于粒子不断破碎,产生许多新的断裂面,此时没有足够的分散剂作用于新产生的断裂面以提供静电空间位阻作用,从而容易发生团聚,因而不利于后续研磨。此外,早期添加的过量分散剂虽然分子结构被破坏不能起到分散的作用,但是仍然留在悬浮液中,这些残余高分子物质的存在会对研磨后GCC悬浮液的流动性产生负面影响。综合分析实验结果,选择分段添加的方式。
2.4 与商品后段分散剂助磨效果比较
表1为P(AA-AMPS)与目前市售的后段分散剂MN-9、LX-7的对比结果。由表1看出,在最佳研磨条件下,与MN-9和LX-7相比,P(AA-AMPS)研磨GCC浆液<2 μm的含量为97.2%,MN-9与LX-7分别为94.9%和95.4%,P(AA-AMPS)具有更好的助磨效果。
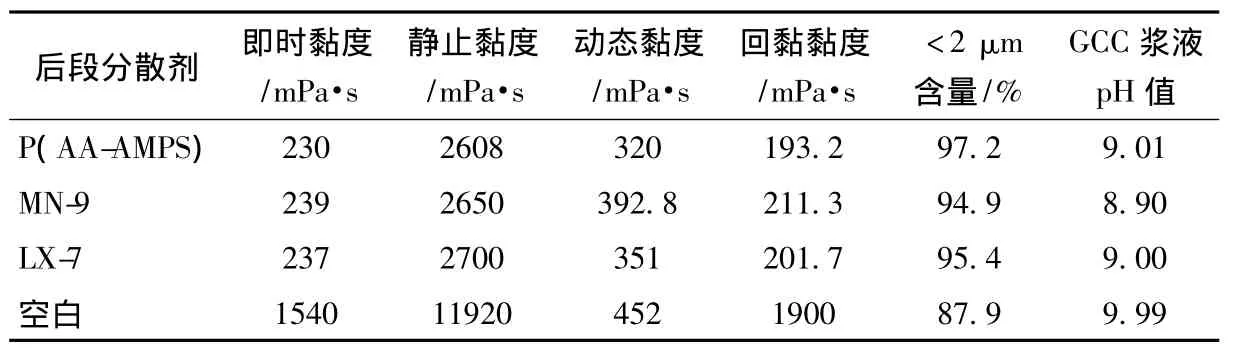
表1 不同后段分散剂对GCC浆液助磨效果的影响
3 结论
采用水溶液自由基聚合的方法,以丙烯酸(AA)和2-丙烯酰胺-2-甲基丙磺酸 (AMPS)为聚合单体,过硫酸铵为引发剂,次亚磷酸钠为链转移剂,制备研磨碳酸钙 (GCC)超细研磨用分散剂二元聚合物P(AA-AMPS)。
3.1 通过单因素实验,确定了P(AA-AMPS)的最佳合成工艺为:单体配比m(AMPS)∶m(AA)=0.25、过硫酸铵用量4%(占单体总质量分数)、次亚磷酸钠用量7%(占单体总质量分数)、反应时间5.5 h、反应温度 80℃。
3.2 实验确定了P(AA-AMPS)的最佳使用条件为:分散剂用量1.2%,研磨机转速1000 r/min,采用分段添加的方式。
3.3 在最佳应用条件下,与常用的市售LX-7、MN-9等GCC浆液助磨分散剂相比,P(AA-AMPS)对GCC浆液的助磨效果更明显。
[1] Wang Haisong,Liu Jingang.New development in coating technology[J].World Pulp and Paper,2003,22(5):6.王海松,刘金刚.涂布技术的最新进展[J].国际造纸,2003,22(5):6.
[2] SU Yan-qun,CAO Zhen-lei.Effect of Coating Pigments on Coating Coverage and Paper Properties of LWC[J].China Pulp & Paper,2005,24(2):3.苏艳群,曹振雷.涂布颜料对涂布纸性质和涂层覆盖的影响[J].中国造纸,2005,24(2):3.
[3] SONG Bao-xiang,ZHANG Cheng.Property and Application of Papermaking Grade CaCO3[J].China Pulp & Paper,2005,25(4):54.宋宝祥,张 成.造纸碳酸钙品质特性与应用现状及前景[J].中国造纸,2005,25(4):54.
[4] ZHANG Mei-yun,HU Kai-tang,PING Qing-wei,et al.Processing of paper and specialty paper[M].Version 3.Beijing:China Light Industry Press,2010.张美云,胡开堂,平清伟,等.加工纸与特种纸[M].3版.北京:中国轻工业出版社,2010.
[5] A Kwade,J Schwedes.Breaking characteristics of different materials and their effect on stress intensity and stress number in stirred media mills[J].Powder Technol,2002,122(7):109.
[6] Ji Guangbin,Cal Xiaoli,Chen Wrizhong.Applications and preparative process of ultrafine ground CaCO3[J].Shanghai chemical Industry,2000,25(11):19.姬广斌,柴晓利,陈伟忠.超细重质碳酸钙的应用及制备工艺[J].上海化工,2000,25(11):19.
[7] Xie Xiaosheng,Shi Qiang.Present Development of grinding technology of ultrafine ground calcium carbonate in paper industry[J].China powder science and technology,2004,23(6):40.谢晓生,石 强.造纸用超细重质碳酸钙研磨技术进展[J].中国粉体技术,2004,23(6):40.
[8] Song Baoxiang.Prospect and application of calcium carbonate in the paper making industry[J].Non-Metallic Mines,2001,24(2):5.宋宝祥.碳酸钙在造纸工业中的应用现状及展望[J].非金属矿,2001,24(2):5.
[9] He Jing,Wu Yu-ying;Liu Liu-jun et al.Synthesis of low-molecular sodium polyacrylate and capability of dispersing[J].Journal of Beijing Forestry University,2002,24(5/6):216.何 静,吴玉英,刘六军,等.低分子质量聚丙烯酸钠的合成及分散性能研究[J].北京林业大学学报,2002,24(5/6):216.
[10] LI Jian-wen,TIAN Zhong-jian,QIU Hua-yu.Preparation and Application of the Novel Polyacrylic Acid Sodium as Pigment Dispersant[J].China Pulp & Paper,2004,23(8):10.李建文,田中建,邱化玉.低分子质量聚丙烯酸钠的合成及应用[J].中国造纸,2004,23(8):10.
[11] Dai Q,Wu D Z,Zhang Z,et al.Preparation of monodisperse poly(methyl methacrylate)particles by radiation-induced dispersion polymerization using vinyl terminus polysiloxane macromonomer as a polymerizable stabilizer[J].Polymer,2003,44(1):73.
[12] Hong J,Hong C K,Shim S E.Synthesis of polystyrene microspheres by dispersion polymerization using poly(vinyl alcohol)as a steric stabilizer in aqueous alcohol media[J].Colloids and Surfaces A:Physicochemical and Engineering Aspects,2007,302(1):225.
[13] Okubo T,Takezawa K,Kimura H.Suspension viscosity of colloidal crystals and liquids in exhaustively deionized aqueous suspensions coexisting with ion-exchange resins[J].Colloid and Polymer Science,2000,278(6):571.
[14] Zhao Rui-ying,Zhao Xin.Synthesis and scale inhibition performance of copolymers of acrylic acid and 2-acrylamido-2-methylpropanesulfonic acid[J].Technology of Water Treatment,2010,36(9):53.赵瑞英,赵 新.丙烯酸与2-丙烯酰胺-2-甲基丙磺酸共聚物的合成及阻垢性能[J].水处理技术,2010,36(9):53.
[15] Weimer M W,Chen H,Giannelis E P,et al.Direct synthesis of dispersed nanocomposites by in situ living free radical polymerization using a silicate-anchored initiator[J].Journal of the American Chemical Society,1999,121(7):1615.
[16] Qiu Kunyuan.Progress of free radical polymerization in recent years[J].Chinese Polymer Bulletin,2008,7:15.丘坤元.自由基聚合近20年的发展[J].高分子通报,2008,7:15.
[17] Matsumura Y,Nakamori T.Steam reforming of methane over nickel catalysts at low reaction temperature[J].Applied Catalysis A:General,2004,258(1):107.
猜你喜欢
选煤技术(2022年2期)2022-06-06
石材(2022年1期)2022-05-23
山东陶瓷(2021年5期)2022-01-17
中学生数理化·中考版(2021年10期)2021-11-22
陶瓷学报(2020年5期)2020-11-09
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
军事文摘(2020年18期)2020-10-27
石材(2020年2期)2020-03-16
世界农药(2019年2期)2019-07-13
新高考·高一物理(2015年6期)2015-09-28

- 中国造纸的其它文章
- 组分间的相互作用对涂料流变性能的影响