
苯甲酰苯并咪唑衍生物的合成
2013-11-19 07:37陈建军周丽萍奚立民叶海伟
合成化学 2013年2期
陈建军, 周丽萍, 奚立民, 叶海伟
(1. 台州职业技术学院 生物与化工学院,浙江 台州 318000; 2. 浙江工业大学 药学院 绿色制药技术与装备教育部重点实验室,浙江 杭州 310014)
苯并咪唑衍生物具有特殊的结构、生理活性及反应活性等,应用十分广泛,可作为药物中间体,在抗癌、抗真菌、消炎镇痛、抗风湿、驱虫等方面有重要的药用价值[1~5]。其对前列腺素D合成酶的活性,特别对由前列腺素D2或其代谢产物介导的疾病如过敏性、炎症等方面具有较好的抑制作用,也是重度阿尔茨海默氏症和大脑损伤等疾病的良好抑制剂[6]。近年来,该类杂环化合物的合成及其生物活性研究日益成为杂环化学研究的热点[7~9]。其合成方法按起始原料分类有多种,其中以邻苯二胺和醛为原料的路线反应条件温和,原料廉价易得,收率较高,但要经历氧化脱氢历程,需2eq以上氧化剂(如对苯醌, Pb(OAc)4, DDQ, MnO2等[5])催化,且产物分离提 纯困难。因此,选择较佳的氧化剂及用量是制备较高收率苯并咪唑衍生物的关键。

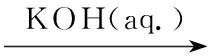
Scheme1
本文在文献[10]方法基础上,以3,5-二甲基-1H-吡咯-2-甲酸乙酯(2)为起始原料,与新型Vilsmeier试剂反应得4-甲酰基-3,5-二甲基-1H-吡咯-2-甲酸乙酯(3);在K2S2O8-CuSO4体系催化下, 3与3,4-二氨基二苯甲酮环合制得到4-(5-苯甲酰-1H-苯并咪唑-2-基)-3,5-二甲基-1H-吡咯-2-甲酸乙酯(4); 4水解后与2-吡唑啉经脱水反应合成了一个苯并咪唑衍生物(1, Scheme 1),总收率54%,其结构经1H NMR,13C NMR和MS确证。
1 实验部分
1.1 仪器与试剂
X-4型显微熔点仪;Varian-400 MHz型核磁共振仪(DMSO-d6为溶剂, TMS为内标);Trace DSQ FINNIGSN型质谱仪。
所用试剂均为分析纯,用前均按标准方法作纯化处理。
1.2 合成
(1) 3的合成
向三颈烧瓶中依次加入双(三氯甲基)碳酸酯(BTC)10.1 g(34 mmol)和1,2-二氯乙烷25 mL,搅拌下冷却至0 ℃,滴加DMF 7.3 g(100 mmol),滴毕,于室温反应30 min;冷却至0 ℃~5 ℃,滴加2 6.7 g(40 mmol)的1,2-二氯乙烷(15 mL)溶液,滴毕,于85 ℃反应2 h。蒸除有机相,冷却至0 ℃,加入饱和碳酸钠溶液40 mL,搅拌2 h(析出固体),过滤,滤饼干燥得棕黄色固体3 7.0 g,收率90%, m.p.141 ℃~143 ℃(140 ℃~142 ℃[11]);1H NMRδ: 12.02(s, 1H), 9.88(s, 1H), 4.23(q,J=7.2 Hz, 2H), 2.46(s, 3H), 2.43(s, 3H), 1.28(t,J=7.2 Hz, 3H);13C NMRδ: 187.6, 164.2, 139.0, 124.9, 123.6, 122.5, 61.3, 14.7, 14.5, 11.9; ESI-MSm/z: 196.2{[M+H]+}。
(2) 4的合成
在三颈烧瓶中依次加入3,4-二氨基二苯甲酮8.4 g(39.5 mmol), 3 7.0 g(35.9 mmol), K2S2O89.7 g(35.9 mmol), CuSO40.6 g(3.6 mmol)和乙醇70 mL,搅拌下于70 ℃反应6 h。冷却至室温,加水40 mL,搅拌(析出固体),过滤,滤饼用水(50 mL)洗涤,乙酸乙酯重结晶得黄色固体4 11.8 g,收率85%, m.p.133 ℃~135 ℃;1H NMR(CDCl3)δ: 9.16(s, 1H), 8.10~8.02(m, 1H), 7.83~7.79(m, 3H), 7.68~7.65(m, 1H), 7.60~7.56(m, 1H), 7.50~7.46(m, 2H), 4.33(q,J=7.2 Hz, 2H), 2.58(s, 3H), 2.55(s, 3H), 1.37(t,J=7.2 Hz, 3H), 1.25(s, 1H);13C NMR(CDCl3)δ: 195.6, 162.5, 140.1, 138.5, 137.9, 136.2, 136.0, 132.5, 132.1, 131.7, 128.9(C2), 128.1(C2), 122.5, 120.5, 111.6, 110.3, 108.1, 61.5, 14.4, 13.6, 12.3; ESI-MSm/z: 388.2{[M+H]+}。
(3) 5的合成
在三颈烧瓶依次加入乙醇100 mL, 4 6.0 g(15.5 mmol),搅拌使其溶解,滴加氢氧化钾71.4 mmol溶液25 mL,滴毕,于75 ℃反应5 h。冷却至室温,用1 mol·L-1盐酸调至中性,析出固体,过滤,滤饼用丙酮(20 mL)洗涤,减压干燥得淡黄色固体5 5.0 g, 收率90%, m.p.236 ℃~238 ℃;1H NMRδ: 11.77(s, 1H), 7.94~7.90(m, 1H), 7.75~7.66(m, 5H), 7.59~7.55(m, 2H), 3.15(s, 1H), 2.50(s, 3H), 2.49(s, 3H), 1.35(s, 1H);13C NMRδ: 195.8, 162.7, 139.8, 138.1, 137.9, 136.3, 136.0, 132.3, 132.1, 128.7(C2), 128.1(C2), 123.5, 121.5, 120.7, 111.6, 110.3, 102.1, 15.2, 14.3; ESI-MSm/z: 360.2{[M+H]+}。
(4)1的合成
在三颈烧瓶依次加入2-吡唑啉1.9 g(27.8 mmol),1-羟基苯并三氮唑(HOBt)2.8 g(20.9 mmol), 1-乙基-3(3-二甲基丙胺)-碳二亚胺(EDCI)4.0 g(20.9 mmol), 5 5.0 g(13.9 mmol)和DMF 70 mL,搅拌于室温反应24 h。加水50 mL,用二氯甲烷(3×50 mL)萃取,合并有机相,依次用饱和碳酸钠溶液, 1 mol·L-1盐酸30 mL,饱和食盐水30 mL洗涤,无水Na2SO4干燥,减压蒸除溶剂后用丙酮重结晶得淡黄色固体14.5 g, 收率78%, m.p.146 ℃~148 ℃(148 ℃~150 ℃[6]);1H NMR (CDCl3)δ: 10.56(s, 1H), 7.99~7.97(m, 1H), 7.79~7.70(m, 3H), 7.57~7.42(m, 4H), 7.27(s, 1H), 7.00(t,J=6.8 Hz, 1H), 3.92(t,J=10.0 Hz, 2H), 2.83(t,J=10.0 Hz, 2H), 2.57(s, 3H), 2.43(s, 3H);13C NMR(CDCl3)δ: 196.7, 157.8, 151.4, 147.6, 138.2(C2), 132.3, 131.6, 131.1, 129.7(C2), 129.1, 127.9(C2), 124.4(C2), 119.3(C2), 112.7(C2), 42.6, 31.4, 12.7, 11.8; ESI-MSm/z: 412.2{[M+H]+}。
2 结果与讨论
BTC参与的反应条件温和,收率较高,既安全又方便,可作为光气、草酰氯、氯化亚砜等的理想替代品,在医药化工中间体的合成中有极为广泛的应用[12]。本文考察了不同Vilsmeier试剂对3收率的影响,结果见表1。由表1可见当Vilsmeier试剂为BTC/DMF时,3的收率最高(90%)。按Vilsmeier试剂活性中间体反应活性次序[13~14](Chart 1),理论上(CF3SO2)2O/DMF形成的Vilsmeier试剂甲酰化活性最高,3的收率应最高,结果恰好相反,原因可能是(CF3SO2)2O/DMF甲酰化反应活性最高,但形成的活性中间体不稳定,选择性反而差,副产物增多从而导致收率较低。 BTC/DMF甲酰化活性最低,但形成的活性中间体稳定,选择性好,收率最高。
以BTC/DMF为Vilsmeier试剂,考察r[n(Vilsmeier试剂) ∶n(2)]对3收率的影响,结果见表2。由表2可见,当r=2.5时,3收率最高(90%)。

*r=n(Vilsmeier试剂) ∶n(2)=2.5,其余反应条件同1.2(1)

表 2 r对3收率的影响*Table 2 Effect of r on the yield of 3
*r=n(Vilsmeier试剂) ∶n(2),其余反应条件同1.2(1)

表 3 氧化剂及其用量对4收率的影响*Table 3 Effect of oxidant and usage amount on the yield of 4
*q=n(氧化剂) ∶n(3),a100 ∶1,其余反应条件同1.2(2)
在4的合成中,重点考察了Na2S2O5, DDQ, MnO2等常见氧化剂及其用量对其收率的影响,结果见表3。由表3可见, K2S2O8-CuSO4催化体系用量最少,后处理较为方便, 4收率最高。
3 结论
本文报道了一种合成苯并咪唑衍生物的新方法,该方法具有反应条件温和、操作简便、反应时间短、收率高、对环境相对友好等优点,有较好的应用前景。
[1] Craigo W A, Lesueur B W, Skibo E B. Design of highly active analogues of the pyrrolo[1,2-a]benzimidazole antitumor agents[J].J Med Chem,1999,42(17):3324-3333.
[2] Gudmundsson K S, Tidwell J, Lippa N,etal. Synthesis and antiviral evaluation of halogenatedβ-D-and-L-erythrofu ranosyl-benzimidazoles[J].J Med Chem,2000,43(12):2464-2472.
[3] Dekhane D V, Pawar S S, Gupta S V,etal. Lithium bromide catalyzed solvent free method for synthesis of 2-substituted benzimidazoles and imidazopyridines[J].Chin Chem Lett,2010,21(5):519-523.
[4] Hirashima S, Suzuki T, Ishida T,etal. Benzimidazole derivatives bearing substituted biphenyls as hepatitis C virus NS5B RNA-dependent RNA polymerase inhibitors:Structure-activity relationship studies and identification of a potent and highly selective inhibitor JTK-109[J].J Med Chem,2006,49(15):4721-4736.
[5] Lin S N, Yang L H. A simple and efficient procedure for the synthesis of benzimidazoles using air as the oxidant[J].Tetrahedron Lett,2005,46(25):4315- 4319.
[6] Yoshihiro U, Yoshiki T, Keiko Y,etal. Benzoimidazole compound capable of inhibiting prostaglandin D synthetase[P].WO 200 700 777 8,2007.
[7] Lin S Y, Isome Y, Stewart E,etal. Microwave-assisted one step high-throughput synthesis of benzimidazoles[J].Tetrahedron Lett,2006,47(17):2883- 2886.
[8] Vázquez G N, Diaz H M, Crespo F A,etal. Design,microwave-assisted synthesis,and spasmolytic activity of 2-(alkyloxyaryl)-1H-benzimidazole derivatives as constrained stilbene bioisosteres[J].Bioorg Med Chem Lett,2006,16: 4169-4173.
[9] 李焱,马会强,王玉炉. 苯并咪唑及其衍生物合成与应用研究进展[J].有机化学,2008,28:210-217.
[10] Kumar A, Maurya R A, Saxena D. Diversity-oriented synthesis of benzimidazole,benzoxazole,benzothia-zole and quinazolin-4(3H)-one libraries via potassium persulfate-CuSO4-mediated oxidative coupling reactions of aldehydes in aqueous micelles[J].Mol Divers,2010,14:331-341.
[11] Kumar S, Kumar D. Syntheses and characterization of pyrrole containing hydrazones[J].Organic Chemistry:An Indian Journal,2011,7(3):148-154.
[12] Su W K, Weng Y Y, Zheng C,etal. Recent developments in the use of bis-(trichloromethyl) carbonate in synthesis[J].Org Prep Proced Int,2009,41(2):93-141.
[13] Alunni S, Linda P, Marino G,etal. Mechanism of the Vilsmeier-Haack reaction.Ⅱ.Kinetic study of the formylation of thiophene derivatives with dimethylformamide and phosphorus oxychloride or carbonyl chloride in 1,2-dichloroethane[J].J Chem Soc,Perkin Transactions 2,1972,(14):2070-2073.
[14] Tebby J C, Willetts S E. Structure and reactivity of the Vilsmeier formylating reagent[J].Phosphorus Sulfur,1987,30:293-295.
猜你喜欢
中国饲料(2021年17期)2021-11-02
中国资源综合利用(2017年2期)2018-01-22
益寿宝典(2017年1期)2017-09-03
广东饲料(2016年5期)2016-12-01
中小学实验与装备(2016年1期)2016-04-19
合成化学(2015年2期)2016-01-17
合成化学(2015年10期)2016-01-17
红领巾·探索(2014年8期)2014-10-10
应用化工(2014年1期)2014-08-16
无机化学学报(2014年12期)2014-02-28
