
大分子自由基加成交叉偶合反应制备嵌段共聚物及其表征
2013-11-18 08:23何奕虹
化学反应工程与工艺 2013年6期
何奕虹,王 齐
(高分子合成与功能构造教育部重点实验室 浙江大学高分子科学与工程学系,浙江 杭州 310027)
嵌段聚合物的合成方法主要有两种:一是逐段法,即通过阴离子聚合[1,2]或者活性可控自由基聚合的手段,如原子转移自由基聚合(ATRP)[3-6]、可逆-加成断裂链转移(RAFT)[7-9]和氮氧自由基调控聚合(NMP)[10,11]等,采用间歇投料的方式,连续引发多种单体聚合得到嵌段聚合物;二是偶联法,即利用含官能团的聚合物之间高效的偶合反应,如Cu(I)催化1,3-二偶极环加成(CuAAC)反应[12-14]、Diels-Alder(DA)反应[15-17]和巯基-烯烃偶合反应[18,19]等,将两个及以上的聚合物连接起来,得到嵌段聚合物。自由基的偶合反应是一个高效反应,Zhang 等[20]报道一种基于自由基加成交叉偶合反应(Radical Addition Cross-Coupling, RACC)的新方法制备两嵌段聚合物,如反应(1)所示,以两种α-溴代聚合物为前体,在Cu 和配体的作用下产生不同的大分子自由基,利用两种自由基与双键化合物的加成速率常数的不同,使一种大分子自由基与双键加成生成中间态自由基,再与另一种大分子自由基偶合,可以合成两嵌段聚合物,该反应过程为自由基的加成-交叉偶合反应(RACC)。
大分子的偶合反应效率主要是通过反应前后平均分子量的变化来表征,如文献[21]用该方法表征单端大分子自由基偶合反应的偶合效率。Zhang 等[20,22]曾经采用凝胶渗透色谱仪(GPC)分峰拟合的方法,获得产物中不同聚合物组分后,根据不同组分的峰面积计算了交叉偶合效率(CCE)。
本工作利用带有紫外检测器的GPC(UV-GPC),研究聚苯乙烯大分子自由基与聚丙烯酸酯大分子自由基的交叉偶合反应,利用苯乙烯单元对紫外光的特定吸收,得到含有苯乙烯单元的组分的GPC图,排除了聚丙烯酸酯大分子对GPC 结果的影响,并结合聚苯乙烯的分子量的选择,可以准确地采用GPC 的结果计算反应的CCE。
1 实验部分
1.1 RACC 反应
实验中用到的试剂有铜粉(粒径3.25~4.75 µm,99.9%,Alfa),N,N,N’,N’,N”-五甲基二乙基三胺(PMDETA,98%,Alfa),1,1,-二苯基乙烯(DPE,98%,Alfa),α-溴代聚苯乙烯(PS-Br),α-溴代聚丙烯酸甲酯(PMA-Br)和四氢呋喃(THF)。其中PS-Br 和PMA-Br 由ATRP 合成[20],THF 经过回流除水后使用。
向Schlenk 瓶中依次加入一定量的DPE,PMA-Br/PS-Br(物质的量之比为1∶1)的THF 溶液(PS-Br的溶度为0.05 mol/L),PMDETA,混合后经冷冻-脱气-熔融循环3 次,再于液氮中冷冻,并在氮气氛围下加入一定量铜粉,密封后抽真空-通氮气3 次,室温下解冻后置于设定温度反应一定时间,接触空气停止反应。粗产物过中性氧化铝柱除去铜盐,CH2Cl2/CH3OH(体积比25∶1)为淋洗剂,浓缩,真空干燥过夜。
1.2 表征方法
聚合物的数均分子量(Mn)及其分子量分布(PDI)在Waters 1525/2414 凝胶渗透色谱仪上测定,采用3 根PLgel 10 µm 的柱子,孔径分别为50,100 和1 000 nm,检测器为示差(RI)和紫外(UV/254 nm)两种检测器,流动相为THF,流速为1.0 mL/min,检测温度为30 ℃,检测样品的质量分数为3‰。1H-NMR 在Bruker DMX-400 MHz 的核磁仪上测定,以氘代氯仿(CDCl3)为溶剂,四甲基硅烷(TMS)为内标,室温下测试。
2 结果与讨论
2.1 不同检测器的GPC 结果比较
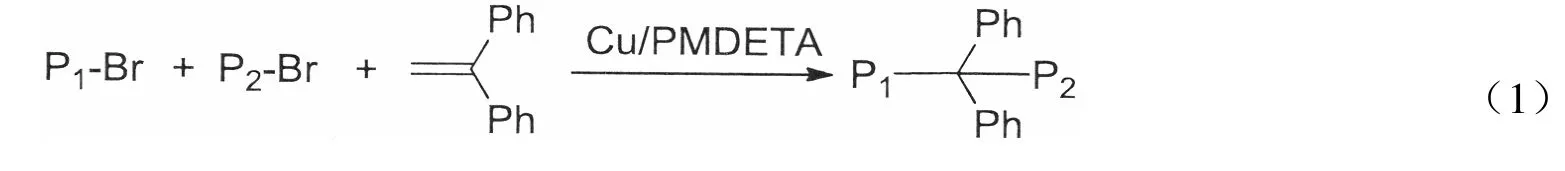
比较了示差和紫外两种检测器下RACC 反应前体PMA-Br/PS-Br(物质的量之比为1∶1)的GPC曲线,如图1所示。结果发现,示差检测器下的GPC曲线(RI-GPC)中PS-Br 和PMA-Br 的信号都很强,紫外检测器下的UV-GPC 曲线中PMA 的峰面积比例只有3.3%,对GPC 曲线峰面积的贡献几乎可以忽略。这主要是因为PS 对254 nm 的紫外光具有较强的吸收,而PMA 对254 nm 的紫外光几乎没有吸收,故UV-GPC 曲线只反映了含有PS 单元的聚合物组分。
将RACC 产物用GPC 的示差和紫外两种检测器分别进行检测,对得到的RI-GPC 曲线和UV-GPC 曲线进行分峰拟合,采用高斯函数拟合不同组分的聚合物的分子量分布,结果如图2所示。
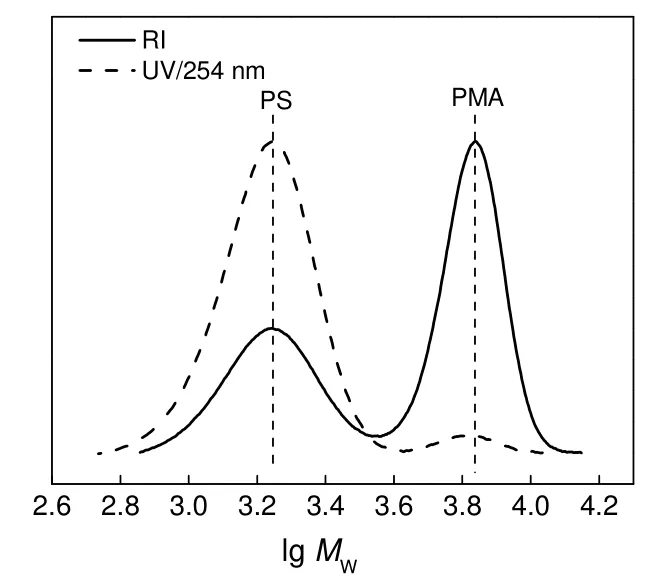
图1 PMA-Br/PS-Br(1/1, mol/mol)的RI-GPC 曲线和UV-GPC 曲线Fig.1 GPC curves of PMA-Br/PS-Br(1/1,mol/mol)
如图2(a)所示,整个UV-GPC 曲线包含了3 个明显的峰。根据峰顶分子量的大小,可以判断:第一个峰的分子量与原料PS-Br 分子量相近,为单段PS,可能来源于未反应的原料或岐化和单基终止等复杂反应;第二个峰的分子量为原料分子量的近2 倍,是原料PS-Br 的自偶合产物PS-PS;第三个峰的分子量接近PS-Br 和PMA-Br 的分子量之和(Mn=MnPMA+MnPS+MnDPE-2MnBr,Mn为8 130 g/mol),判断其为交叉偶合产物PMA-DPE-PS。根据PMA-DPE-PS 在总产物GPC 曲线中所占峰面积的比例,即可得到基于PS 的CCE。粗产物的各组分在UV-GPC 曲线中直观明了,分峰简单,计算CCE简便快速。
在RACC 反应中,选取了分子量较小的PS-Br 和分子量较大的PMA-Br 作为反应前体,使得产物中含有PS 的3 种组分在UV-GPC 曲线中区分明显。若选取分子量接近的PS-Br 和PMA-Br 进行RACC,自偶合产物PS-PS 和交叉偶合产物PMA-DPE-PS 的分子量接近,在UV-GPC 曲线中难以被区分;若选取分子量较大的PS-Br 和分子量较小的PMA-Br 进行RACC,单段PS 和交叉偶合产物PMA-DPE-PS的分子量接近,在UV-GPC 曲线中难以被区分。
RI-GPC 曲线反映的不只是PS 部分,也包括PMA 部分,PMA 自由基倾向于岐化终止,故分峰拟合中将未转化为嵌段聚合物的PMA 部分理解为只有单段PMA。如图2(b)所示,RI-GPC 曲线的组分构成相对复杂,且各组分在RI-GPC 曲线中不明显,故用RI-GPC 曲线计算CCE引入的误差比用UV-GPC 曲线大,故下面采用UV-GPC 来计算CCE。

图2 PMA-Br 和PS-Br 的RACC 粗产物的GPC 曲线及高斯分峰拟合结果Fig.2 GPC curves of the crude product of RACC from PMA-Br and PS-Br and its Gaussian fitting result
2.2 反应条件对CCE 的影响
在RACC 反应中,Cu 将溴代基团还原产生自由基。不同[Cu]0/[PS-Br]0比对RACC 反应的影响如图3和表1所示。由图和表可知,随着[Cu]0/[PS-Br]0比例的增加,产物中单段PS 的含量由72.1%减小到16.6%,PS-PS 的含量由 0 增加到 16.7%,PMA-DPE-PS 的含量和CCE由27.9%增大到66.7%。改变[Cu]0/[PS-Br]0比例引起这些变化的原因是:当Cu(0)/PMDETA 的浓度较低时,大分子自由基产生的速率较慢,影响了大分子自由基的反应速率,使得反应1 h 的产物中存在较多未反应的PS-Br,相应交叉偶合产物也比较少,CCE不高。当Cu(0)/PMDETA的浓度增加时,大分子自由基产生的速率增大,相同的反应时间内前体PS-Br 消耗得多,生成的PS-PS和PMA-DPE-PS 也较多,CCE相应增加。
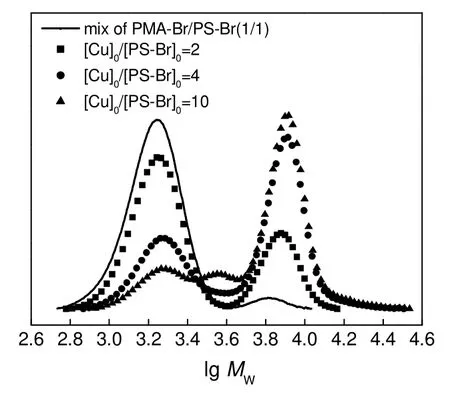
图3 不同[Cu]0/[PS-Br]0 投料比下PS-Br/PMA-Br(1/1,mol/mol)及其RACC 粗产物的UV-GPC 曲线Fig.3 UV-GPC curves of the mixture of PMA-Br/PS-Br(1/1,mol/mol) and the crude coupling products under different[Cu]0 /[PS-Br]0 ratios

表1 不同条件下RACC 粗产物的分析结果Table 1 Results of the crude products of RACC of PS-Br and PMA-Br1) under different conditions
比较[Cu]0/[PS-Br]0分别为4∶1 和10∶1 时产物中PS-PS 的含量,前者为7.3%,后者为16.7%,这是由于铜加入量较多时,产生大分子自由基的速率快,造成PS 自由基的浓度较高,导致了PS 自由基的自偶合反应。
1,1-二苯基乙烯(DPE)在反应中的作用是与大分子自由基进行加成反应,将其转变成为更加稳定的大分子自由基。[DPE]0/[PS-Br]0对RACC 反应的影响如图4和表2所示。由图和表可知,当[DPE]0/[PS-Br]0由1∶1 增大到2∶1 时,产物中单段PS 的含量减小了11.2%,PS-PS 的含量变化不大,只有1.3%的降低,PMA-DPE-PS 的含量和CCE 增大约11%,增幅明显。这是因为DPE 的用量增加后,产生的PMA 自由基容易被过量的DPE 捕捉,其自身的岐化终止等副反应能有效被抑制,有利于与PS 自由基发生交叉偶合反应,同时抑制PS 自由基发生岐化或单基终止等副反应。当[DPE]0/[PS-Br]0由2∶1 增大到5∶1 时,单段PS,PS-PS 和PMA-DPE-PS 的含量变化不明显,变化最小为0.7%,最多为2.2%,CCE只增加了约2%。这是因为在[DPE]0/[PS-Br]0为2:1 的情况下,DPE 对前体聚合物来讲已是过量,此时再增加DPE 的用量对增大CCE起的作用已经不明显了,故[DPE]0/[PS-Br]0为2∶1 比较合适。
表2 不同条件下RACC 粗产物的分析结果Table 2 GPC results of the crude products of RACC of PS-Br and PMA-Br1) under different [DPE]0 /[PS-Br]0 ratios and the Gaussian fitting result of signals of UV/254 nm detector
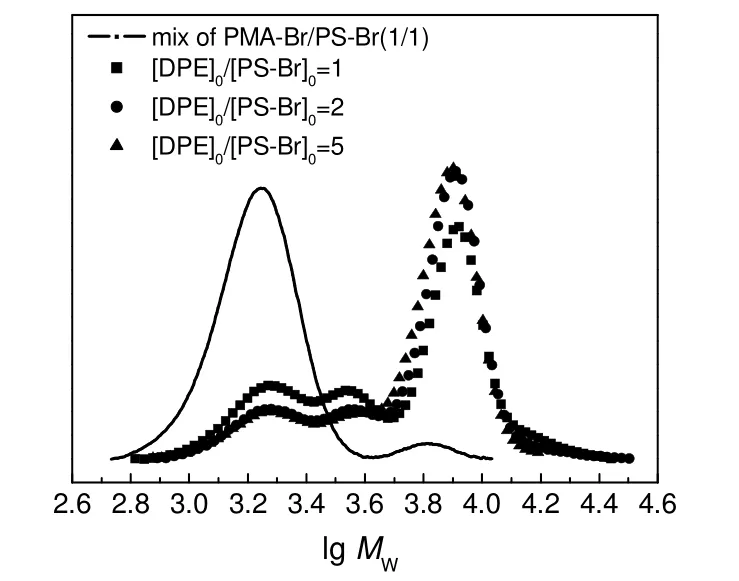
图4 不同[DPE]0/[PS-Br]0 下PS-Br/PMA-Br(1/1, mol/mol)及其RACC 粗产物的UV-GPC 曲线Fig.4 UV-GPC curves of the mixture of PMA-Br/PS-Br(1/1,mol/mol) and the crude coupling products under different[DPE]0 /[PS-Br]0 ratios
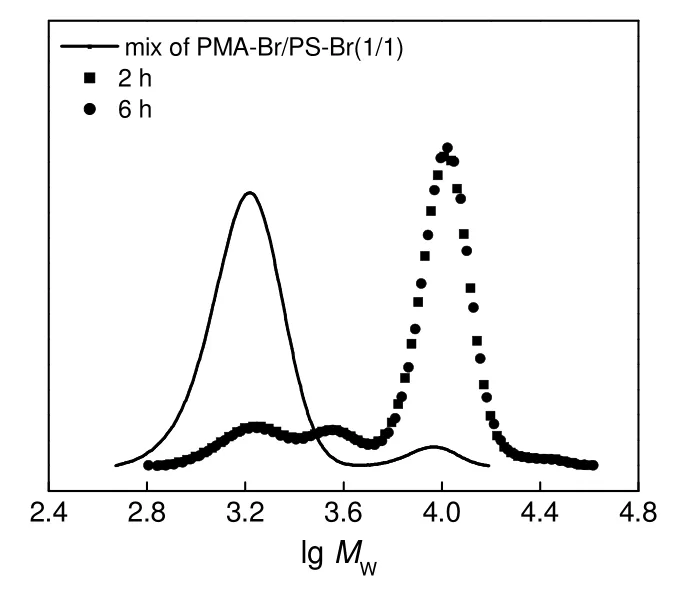
图5 不同反应时间下PS-Br/PMA-Br(1/1, mol/mol)及其RACC 粗产物的 UV-GPC 曲线Fig.5 UV-GPC curves of the mixture of PMA-Br/PS-Br(1/1,mol/mol) and the crude coupling products under different reaction time
反应时间对RACC 反应的影响如图5和表3所示。反应2 h 和6 h 的产物的GPC 曲线吻合较好,其中各个组分的含量,无论是单段PS,PS-PS 或是PMA-DPE-PS,均基本相同,CCE分别为76.9%和78.1%,故反应2 h 和6 h 的结果相差不大,可以推测反应2 h 时RACC 已经达到反应终点。

表3 不同反应时间下PS-Br 和PMA-Br 的RACC 粗产物的UV- GPC 信号和高斯分峰拟合结果Table 3 GPC results of the crude products from RACC of PS-Br and PMA-Br1) under different reaction time and the Gaussian fitting result
表3和表2中,[Cu]0/[PS-Br]0分别为4∶1 和10∶1,其单段PS 的含量差不多,但前者的PS-PS 含量相对小,CCE相对高。这是因为当[Cu]0/[PS-Br]0为10∶1 时,催化剂浓度过高,产生了较多PS 自由基,导致自偶合副产物PS-PS 增多,CCE相对不高,减小 [Cu]0/[PS-Br]0为4∶1 时,PS 自由基产生速率降低,PS-PS 的含量减小,幅度超过5%,CCE从70%左右提高到75%以上,最高为78.1%。
将反应2 h 的产物进行1H-NMR 表征,如图6所示。前体PS-Br 中与端基溴相连碳上氢原子的化学位移在(4.32~4.57)×10-6(4.32~4.57 ppm)处,PMA-Br中与端基溴相连的碳上的氢原子的化学位移在(4.21~4.29)×10-6(4.21~4.29 ppm)处,当RACC反应了2 h 后,以上两个峰在生成的粗产物的1H-NMR 谱图中均没有发现,说明PS-Br 和PMA-Br 溴端基已完全转化,反应2 h 时RACC 已经达到反应终点。

图6 PMA-Br(c),PS-Br(b)及其自由基加成偶合粗产物(a)的1H-NMR(CDCl3, 400 MHz)图谱Fig.6 1H-NMR(CDCl3, 400 MHz) spectra of PMA-Br(c),PS-Br (b) and their crude product(a) prepared by RACC
3 结 论
选择适当分子量的PMA-Br 和PS-Br 作为RACC 反应前体,用带有紫外检测器的GPC 表征产物,提出了一种计算CCE的新方法。采用该方法计算得到的CCE相比由RI-GPC 曲线得到的CCE更加接近实际值。新表征方法的应用为进一步改进RACC 条件提供重要的检测和分析手段。
致 谢
感谢国家自然科学基金(21174123)的资助。
[1]Feldthusen J, Iván B, Müller A H E.Synthesis of linear and star-shaped block copolymers of isobutylene and methacrylates by combination of living cationic and anionic polymerizations[J].Macromolecules, 1998, 31(3):578-585.
[2]Ramireddy C, Tuzar Z, Prochazka K, et al.Styrene-tert-butyl methacrylate and styrene-methacrylic acid block copolymers:synthesis and characterization[J].Macromolecules, 1992, 25(9):2541-2545.
[3]Can A, Altuntas E, Hoogenboom R, et al.Synthesis and maldi-tof-ms of ps-pma and pma-ps block copolymers[J].European Polymer Journal, 2010, 46(9):1932-1939.
[4]Coca S, Paik H J, Matyjaszewski K.Block copolymers by transformations of living ring-opening metathesis polymerization controlled/“living” atom transfer radical polymerization[J].Macromolecules, 1997, 30(21):6513-6516.
[5]Soeriyadi A H, Boyer C, Nyström F, et al.High-order multiblock copolymers via iterative cu(0)-mediated radical polymerizations(set-lrp):toward biological precision[J].Journal of the American Chemical Society, 2011, 133(29):11128-11131.
[6]Davis K A, Matyjaszewski K.Abc triblock copolymers prepared using atom transfer radical polymerization techniques[J].Macromolecules, 2001, 34(7):2101-2107.
[7]Quemener D, Davis T P, Barner-Kowollik C, et al.Raft and click chemistry:a versatile approach to well-defined block copolymers[J].Chemical Communications, 2006, (48):5051-5053.
[8]Luo Y, Liu X.Reversible addition–fragmentation transfer(raft) copolymerization of methyl methacrylate and styrene in miniemulsion[J].Journal of Polymer Science Part A:Polymer Chemistry, 2004, 42(24):6248-6258.
[9]Yang C, Cheng Y L.Raft synthesis of poly(n-isopropylacrylamide) and poly(methacrylic acid) homopolymers and block copolymers:kinetics and characterization[J].Journal of Applied Polymer Science, 2006, 102(2):1191-1201.
[10]Zhang C, Lessard B, Maric M.Synthesis and characterization of benzyl methacrylate/styrene random copolymers prepared by nmp[J].Macromolecular Reaction Engineering, 2010, 4(6-7):415-423.
[11]Savelyeva X, Lessard B H, Marić M.Amphiphilic poly(4-acryloylmorpholine)/poly[2-(n-carbazolyl)ethyl acrylate]random and block copolymers synthesized by nmp[J].Macromolecular Reaction Engineering, 2012, 6(5):200-212.
[12]Kempe K, Krieg A, Becer C R, et al.“Clicking” on/with polymers:a rapidly expanding field for the straightforward preparation of novel macromolecular architectures[J].Chemical Society Reviews, 2012, 41(1):176-191.
[13]Schmidt U, Zehetmaier P C, Rieger B.Direct synthesis of poly(dimethylsiloxane) copolymers with tpe-properties via cuaac (click chemistry)[J].Macromolecular Rapid Communications, 2010, 31(6):545-548.
[14]Lammens M, Fournier D, Fijten MWM, et al.Star-shaped polyacrylates:highly functionalized architectures via cuaac click conjugation [J].Macromolecular Rapid Communications, 2009, 30(23):2049-2055.
[15]Durmaz H, Colakoglu B, Tunca U, et al.Preparation of block copolymers via diels alder reaction of maleimide- and anthracene-end functionalized polymers[J].Journal of Polymer Science Part A:Polymer Chemistry, 2006, 44(5):1667-1675.
[16]Kwart H, King K.The reverse diels-alder or retrodiene reaction[J].Chemical Reviews, 1968, 68(4):415-447.
[17]Dag A, Durmaz H,Tunca U, et al.Multiarm star block copolymers via diels-alder click reaction[J].Journal of Polymer Science Part A:Polymer Chemistry, 2009, 47(1):178-187.
[18]Li M, De P, Gondi S R, et al.End group transformations of raft-generated polymers with bismaleimides:functional telechelics and modular block copolymers[J].Journal of Polymer Science Part A:Polymer Chemistry, 2008, 46(15):5093-5100.
[19]Lowe A B.Thiol-ene “click” reactions and recent applications in polymer and materials synthesis[J].Polymer Chemistry, 2010, 1(1):17-36.
[20]Zhang C, Wang Q.Block copolymers prepared by polymeric radical addition cross-coupling reaction to different double bonds[J].Journal of Polymer Science Part A:Polymer Chemistry, 2013, 51(13):2817-2823.
[21]Sarbu T, Lin K Y, Ell J, et al.Polystyrene with designed molecular weight distribution by atom transfer radical coupling[J].Macromolecules, 2004, 37(9):3120-3127.
[22]Zhu Q, Wang Q.Thermodegradable multisegmented polymer synthesized by consecutive radical addition-coupling reaction ofα,ω-macrobiradicals and dithioester[J].Journal of Polymer Science Part A:Polymer Chemistry, 2012, 50(10):2029-2036.
猜你喜欢
科学家(2022年4期)2022-05-10
油气·石油与天然气科学(2021年9期)2021-10-10
冶金动力(2020年12期)2021-01-04
爆炸与冲击(2019年8期)2019-09-25
中学生物学(2019年1期)2019-01-13
中国社区医师(2018年8期)2018-07-25
腐蚀与防护(2017年1期)2017-05-09
中国神经免疫学和神经病学杂志(2017年5期)2017-01-12
浙江大学学报(理学版)(2016年6期)2016-12-15
山东工业技术(2015年24期)2015-12-10
