
Au/Fe2O3-MOx催化剂在气体纯化中的应用研究
2013-10-17 06:57:26张凤利
低温与特气 2013年4期
张凤利
(1.福州市产品质量检验所,福州 350025;2.国家化学工业气体产品质量监督检验中心,福州 350008)
当今,信息化水平已经成为衡量一个国家综合国力的重要标志。作为信息化支柱的电子工业已发展成为当今世界的战略性工业。而电子元器件的制造基础是超纯、高纯电子化学气体,半导体器件性能的好坏,在很大程度上取决于所用电子气体的质量,电子气体纯度每提高一个数量级,都会极大地推动半导体器件质的飞跃[1]。在集成电路的刻蚀和清洗过程中,电子气体中百万分之几的微量杂质气体进入工序就能导致质量下降,使每个元件的信息量减少,从而使高密度集成电路产品的不合格率增加[1-3]。因此在制备高纯度的气体过程中,纯化工艺显得尤为重要。
高纯永久性气体主要包括高纯 H2、O2、N2、Ar、He,主要是由工业级气体经纯化获得。H2可以采用钯管扩散、低温净化的方法获取,但钯管扩散不能在高压下充瓶;He、Ar可以通过Zr-Al或Zr-V-Fe合金吸气的工艺提纯,过高的提纯温度,在高压下进行净化充装,从安全方面讲应该充分考虑反应器的结构和强度的计算。催化剂在制备高纯气体中发挥了重要作用。工业级永久性气体中主要含有微量H2、O2、N2、CH4、CO 和 CO2等杂质,其纯化而获得高纯气主要是使用催化剂,将CO和H2氧化成CO2和H2O(2CO+O2=2CO2,2H2+O2=2H2O),然后再用分子筛吸附。氮气和氩气的纯化使用氧净化剂和水汽吸收剂。传统的氧净化剂使用还原铜催化剂;水汽和二氧化碳通常是用活性分子筛来捕获。在某些情况下,使用氧化铜催化剂将CO和H2氧化成CO2和H2O,然后再用分子筛吸附。氢气的纯化与氮气类似,均使用催化剂。典型的是用铂催化剂将O2氧化为H2O,然后用分子筛净化除去[4]。由此可见,氧化反应是制备高纯气体纯化过程中的重要环节,氧化反应催化剂性能的好坏直接决定了气体中杂质的脱除效率,因此获得高性能氧化反应催化剂成为气体纯化工艺中研究的重点。
当前氧化反应催化剂研究主要锁定在负载型金属催化剂的研究。Pt、Ru、Pd、Co、Cu、Ag、Fe、Mo 和Au等金属或金属的氧化物对CO吸附能为20~50 kcal/mol,具有中等程度的吸附能力,能够促进CO的吸附和氧化,成为活性组分研究考察的对象;Fe2O3、La2O3、V2O3、CeO2、ZrO2等具有氧化还原的性质,可产生介稳态吸附氧原子,氧化被吸附的CO并放出氢。其中Pt系(PGM)和Au等负载型贵金属催化剂是近10年来的研究热点,但研究发现:PGM/CeO2体系催化剂在富氢气氛下,由于Pt系金属与载体CeO2间存在强相互作用,使得载体CeO2被不可逆还原,催化剂在250℃下就出现线性劣化失活[5],这个问题同样制约了以Pt系金属为活性组分的其它负载型催化剂在纯化器中的应用。而负载型Au催化剂中Au与载体间的相互作用较为适中,且Au资源较为丰富,在价格上具有一定的优势,相对于Pt系催化剂更有研究前景[6]。基于Fe2O3本身具有氧化还原性能,而CeO2具有独特的快速价态调变性质,常作为电子助剂、结构助剂和共催化剂应用于许多催化剂体系[7]。我们的前期工作主要围绕着以Au为活性组分,开展Au/Fe2O3、Au/CeO2等系列催化剂的CO氧化性能研究。Au/Fe2O3催化剂在0~100℃温区具有较高的CO转化率,但选择性较低;通过适宜的制备方法可以制得CO选择性氧化性能与金铁催化剂相当的Au/CeO2-ZrO2催化剂;通过对Au/ZSM和Au/ZSM-Fe2O3催化剂的性能对比表明,催化剂的活性是Au与Fe2O3相互作用的结果;对Fe2O3/ZSM和纯Fe2O3催化剂的稳定性考察表明Fe2O3也会发生失活,这些暗示Au/Fe2O3催化剂的失活可能不仅跟Au有关,跟载体发生的某种变化也有关。通过我们的初步研究,认为该类催化剂低温活性和稳定性的提高还具有一定的潜力,有望通过进一步的改性研究,使之最终应用于制备高纯气体纯化器中。
尽管对这类催化剂的失活机制有一定的共识,如Au/Fe2O3、Au/CeO2这两种负载型Au催化剂的失活主要是由于积炭引起。然而,如何提高催化剂的稳定性,使之满足制备高纯气气体纯化器使用的寿命要求,还需投入大量的工作。
因此,本申请课题拟在前期工作基础上,针对Au/Fe2O3体系的失活机制,解决制约其在气体纯化器应用的催化剂稳定性问题。通过助剂的选择和添加对催化剂进行改性是提高催化剂结构稳定性的有效途径之一。迄今为止,对于Au/Fe2O3催化剂制备工艺的研究多有报道,但针对助剂改善Au/Fe2O3催化剂体系稳定性的研究还不够系统。
本论文中引入了第五周期过渡金属、稀土金属,以考察这些助剂对催化剂性能的影响规律,以期筛选出适宜助剂,使催化剂能保持较高催化活性的同时提高稳定性。并通过对样品进行X射线粉末衍射(XRD)、比表面积和孔结构测试、程序升温还原(TPR)表征,以探讨助剂对Au/Fe2O3催化剂低温CO氧化反应性能的调变机理。
1 实验部分
1.1 Au/Fe2O3-MOx催化剂的制备
将一定浓度的Fe(NO3)3和沉淀剂K2CO3溶液并流加入到搅拌中的底液(pH值为8.0的K2CO3溶液)中,将所得沉淀物经洗涤数次后分散到50 mL去离子水中,并用K2CO3溶液调节pH值为8.0,最后在强烈搅拌下逐滴加入HAuCl4溶液,并用K2CO3溶液维持体系pH值不变,将所得沉淀物洗涤至检测不到氯离子为止,再经120℃干燥,最后在300℃下焙烧2 h(升温速率为5℃/min),自然冷却即制得所需催化剂。
按助剂金属M与Fe摩尔比为1∶20引入助剂MOx,添加方式为溶液加入法和粉末加入法。其中溶液加入法是将Fe(NO3)3和助剂盐溶液混合均匀后,采用改性沉积—沉淀法制备样品。粉末加入法则将一定量的粉末助剂分散到100 mL去离子水中进行超声波处理10 min后,获得的乳浊液作为底液,然后将一定浓度的Fe(NO3)3溶液与K2CO3溶液缓慢地并流滴加到底液中,采用改性沉积—沉淀法制备了金含量为0.5%(w/w)(理论添加量)的改性Au/Fe2O3-MOX催化剂。
1.2 催化剂性能的评价
称取约150 mg、80~100目的催化剂放入反应管中,将反应管装到管路中,使样品处于管式炉的恒温区内进行实验。原料气为工业氮,其CO含量为10×10-6;O2含量为100×10-6。
原料气和反应气由华爱GC-9560气相色谱仪的PDHID检测器测得,最低可检测到10×10-9,采用两个气动六通阀控制Porapak Q和5A分子筛色谱填充柱进样时间和转换时间,在60℃下可用于完全分离 H2、O2、N2、CH4、CO 和 CO2,在室温(25 ℃)连续反应120 h,每隔6 h采集一次数据。
空速计算:
空速=每小时的气流量(mL·h-1)/[装填量(g)/密度(g·mL-1)]
接触时间=装填量(mg)/每秒气流量(mL·s-1)
CO的转化率表示如下:
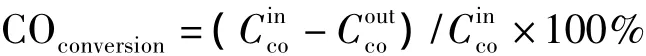
1.3 催化剂的表征
1.3.1 比表面和孔结构测定
采用NOVA 4200e气体吸附孔径测定仪(美国Quantachrome公司)测定催化剂的比表面积和孔结构。样品在120℃抽真空预处理3~4 h后,以N2为吸附质,在液氮温度下(77 K)测得吸脱附曲线。用NOVAWin2/2-P Ver.2.1软件,采用BET法计算比表面积,采用BJH法分析脱附支数据计算孔分布。
1.3.2X射线粉末衍射(XRD)
用玛瑙研钵将所测样品研磨成细微颗粒,然后制成平板状,在荷兰Panalytic公司X’pert Pro衍射仪上采用 X’Celerator探测器,Co靶 Kα(0.1790 nm)辐射,功率 40 kV×40 mA,扫描步长为0.0333 °,扫描速率为40 s·step-1。
1.3.3 H2-程序升温还原(H2-TPR)
采用美国Mciromeritics Autochem 2910型自动吸附仪测定催化剂样品的氧化还原性质。称取催化剂样品0.1000 g,置于玻璃反应器内,在120℃下高纯氦气吹扫1 h,降至室温,继续用高纯氦吹至基线平稳。以10%(V/V)H2/Ar混合气进行程序升温还原至700℃,还原气流30 mL·min-1,升温速率5℃·min-1。
2 结果与讨论
2.1 Au/Fe2O3-MOx催化剂的性能测试
对添加部分第五周期过渡金属、稀土元素的Au/Fe2O3催化剂进行了常温(25℃)下120 h的稳定性评价实验,并与Au/Fe2O3催化剂进行对比,结果如图1所示。所有的催化剂在常温下均具有较高的CO氧化反应初始活性(CO转化率均在100%),但随着反应时间的延长,Au/Fe2O3催化剂的活性逐渐下降,反应120 h后,CO的转化率为95.8%,添加La、Mo和Nb对Au/Fe2O3催化剂稳定性影响较小,稳定性变化不明显,而Ce或Cd的引入则使Au/Fe2O3催化剂稳定性明显下降,值得注意的是含Zr助剂的样品稳定性较未改性的Au/Fe2O3催化剂活性明显改善,经过120 h的反应后,催化剂仍可使CO完全氧化。因此添加ZrO2助剂能有效地提高Au/Fe2O3催化剂的稳定性。
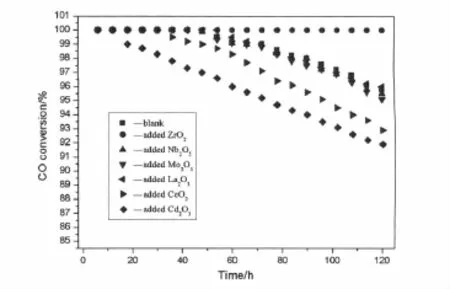
图1 部分第五周期过渡金属、稀土元素对Au/Fe2O3催化剂稳定性的影响Fig.1 Effect of promoter from the fifth periodic transition metal or rare earth element on catalytic stability of Au/Fe2O3catalyst
2.2 织构分析
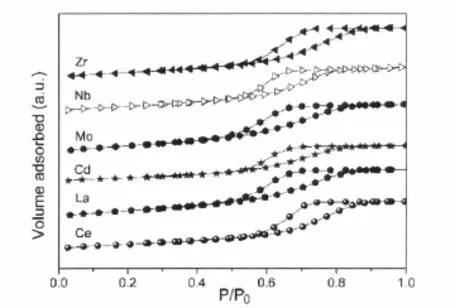
图2 部分第五周期过渡金属、稀土元素改性的Au/Fe2O3-MOx催化剂的等温吸脱附曲线Fig.2 Adsorption-desorption isotherms of Au/Fe2O3 catalysts modified by promoter from the fifth periodic transition metal or rare earth element
图2和表1分别为对该系列样品进行N2物理吸附表征获得的等温吸脱附曲线及比表面积和孔结构具体的分析数据。该系列助剂的引入对催化剂织构影响相对较小。仅发现助剂Mo和La的引入使样品比表面积和孔容有所提高,二者的织构性质相近且活性相当;Zr、Nb、Ce的引入依次使催化剂的比表面积略有下降的同时,增大了样品的孔径和孔容。同样地,通过与该系列样品活性之间关联,可以确认织构性质并不是决定活性优劣的主要原因。
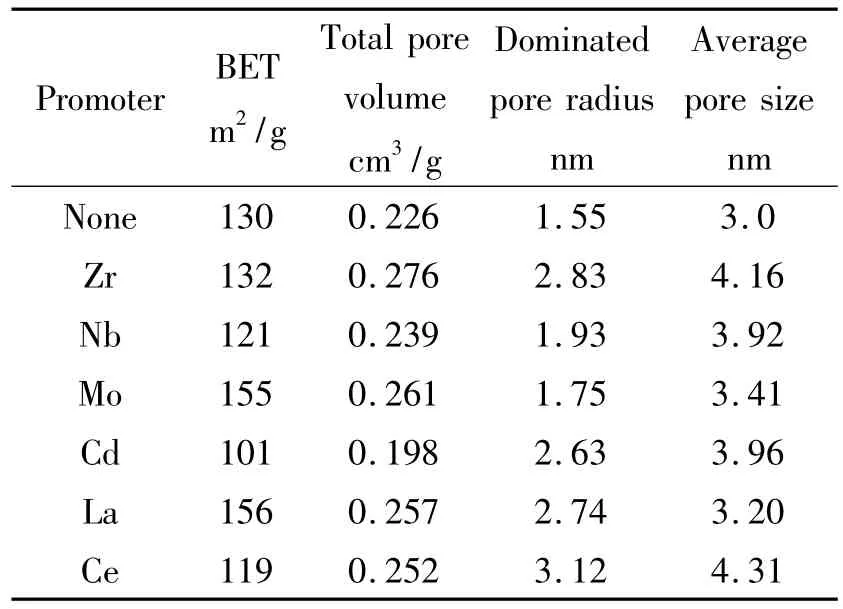
表1 部分第五周期过渡金属、稀土元素对Au/Fe2O3-MOx催化剂织构性质的影响Table 1 Effect of promoter from the fifth periodic transition metal or rare earth element on the texture of Au/Fe2O3-MOxcatalysts
2.3 物相组成分析
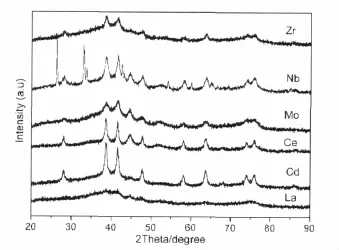
图3 部分第五周期过渡金属、稀土元素改性的Au/Fe2O3催化剂的XRD谱图Fig.3 XRD patters of Au/Fe2O3catalysts modified by promoter from the fifth periodic transition metal or rare earth element
图3中给出了该系列样品的XRD谱图,从中可以明显看出:(1)引入Zr在一定程度上抑制了载体的晶化,提高了金的分散度,这可能是其改善催化剂性能的主要原因。(2)引入Nb后,载体的晶化程度及金的分散状况均没有十分明显的变化;从谱图中能清楚地看到峰形较为尖锐的Nb2O5特征衍射峰,这说明通过粉末加入法引入的Nb2O5主要以晶型完好的独立相存在,其与载体及金的相互作用可以忽略,从而对催化剂的结构和性能影响不大。(3)Mo和La的引入对载体的晶化有明显的抑制作用,特别是La的引入后载体和金的特征衍射峰均明显弱化;而Mo的引入对提高金的分散作用不大。(4)Cd的引入对载体晶化影响不大,但明显提高了金的分散程度;Ce的引入不利于金的分散。通过二者改性后的样品,活性均急剧下降,我们将在下面进行详细的探讨。
2.4 程序升温还原(TPR)分析
对该系列样品进行TPR 表征如图4所示,发现Ce助剂的引入使得原本处于100℃附近归属于表面羟基及无定形氧化铁还原的峰消失,第二个还原峰明显增强并向高温方向移动,由于具有一定活性的载体Fe3O4的形成温度较高,使得样品的催化活性有所下降;同时在300℃附近出现了一个可能与表面Ce还原有关的新还原峰;Cd的引入则使第二个还原峰温向更高温度移动,相应地该样品活性大幅度下降;Nb的引入对催化剂还原性能的影响不明显;而含Zr、Mo和La等助剂的样品由于载体的晶化程度较低,Au的粒子相对较小,第一个还原峰明显宽化,第二个还原峰则向低温方向移动,对应的样品保持了较高的催化活性。
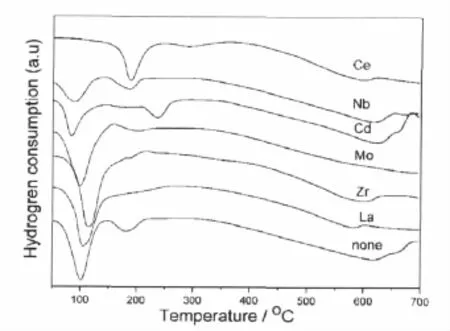
图4 第五周期过渡金属元素改性的Au/Fe2O3催化剂的H2-TPR谱图Fig.4 H2-TPR profiles of Au/Fe2O3catalysts modified by the fifth periodic transition metal promoter
经助剂改性后,催化剂的性能除了与金和载体的物理化学性质有关外,还与助剂本身的性质及助剂与本体催化剂的相互作用有关。引入Ce和Cd后,使得催化剂的还原能力下降,最终导致催化剂性能下降。因此在所考察的Au/Fe2O3催化体系中Ce、Cd不宜作为改性助剂。La和Mo助剂的引入可较大幅度地提高催化剂的比表面积和孔容,但由于前者改性的催化剂中载体的晶化程度较低,而后者改性的催化剂中Au的粒子尺寸较大,使得二者在低温反应时,Au与Fe3O4不能较好地协同作用,导致了低温活性较差。Zr的引入在一定程度上抑制了载体的晶化,并提高了金的分散度,因此具有较高的催化性能。
综上所述,不同助剂对Au/Fe2O3催化剂的还原性能、金的分散度,载体的结晶度产生影响程度不同,使得改性后的Au/Fe2O3催化剂的CO氧化反应活性明显不同,还原性能越好、金的分散度越高,以及适宜的载体结晶度有利于催化剂性能的改善。在所考察的助剂中Zr的引入,明显改善了Au/Fe2O3催化剂的稳定性。
[1]HUSAIN S,KOROS W J.Mixed matrix hollow fiber membranes made with modified HSSZ-1S zeolite in polyetherimide polymer matrix for gas separation [J].Journal of Membrane Science,2007(288):195-207.
[2]MARAND E,PECHAR T W,SANGIL K,et al.Fabrication and characterization of polyimide-zeolite L mixed matrix membranes for gas separations[J].Journal of Membrane Science,2006(277):195-202.
[3]De Vos R M,MAIER W F,VERWEIJ H.Hydrophobic silica membranes for gas separation[J].Journal of Membrane Science,1999(158):277-288.
[4]王书伟,钟惠安.终端气体纯化技术[J].低温与特气,1997,15(1):30-33.
[5]ZALC J M,SOKOLOVSKII V,L?fFLER D G.Are Noble Metal-Based Water-Gas Shift Catalysts Practical for Automotive Fuel Processing[J].Journal of Catalysis,2002(206):169-171.
[6]CAMERONA D,HOLLIDAYB R,THOMPSON D.Gold’s future role in fuel cell systems[J].Journal of Power Sources,2003(118):298-303.
[7]TROVARELLI A,LEITENBURG C,BOARO M,et al.The utilization of ceria in industrials catalysis,Catalysis Today,1999,50(2):353-367.
猜你喜欢
辽宁化工(2022年8期)2022-08-27 06:02:54
建材发展导向(2021年13期)2021-07-28 07:14:40
今日农业(2020年20期)2020-11-26 06:09:20
陶瓷学报(2019年6期)2019-10-27 01:18:38
中国塑料(2016年12期)2016-06-15 20:30:07
当代化工研究(2016年7期)2016-03-20 16:21:53
中国塑料(2015年11期)2015-10-14 01:14:14
中国塑料(2015年9期)2015-10-14 01:12:17
中国塑料(2015年4期)2015-10-14 01:09:19
橡胶工业(2015年8期)2015-07-29 09:22:34
