
碳酸铵水解法制备耐高温介孔CeO2材料
2013-10-17 03:03:10陈山虎史忠华龚茂初陈耀强
无机化学学报 2013年10期
陈山虎 曹 毅 兰 丽 赵 明 史忠华 龚茂初 陈耀强
(四川大学化学学院, 绿色化学与技术教育部重点实验室,成都 610064)
CeO2has attracted much attention in recent years for its applications in many commercial processes,such as catalytic wet oxidation[1],soot combustion[2],and fuel cell technology[3].Moreover,CeO2has also been widely utilized as oxygen storage material in three way catalysts (TWCs)for the purification of exhaust gases from automobile engines[4].CeO2can act as an oxygen buffer by releasing and storing oxygen under rich and lean conditions,respectively,involving the Ce4+/Ce3+couple.This ensures that the air to fuel ratio is kept around the stoichiometric point(14.6),which is crucial to maintain the efficiency of TWCs[5].In addition,multiple functions are also associated with CeO2:stabilization of noble metal dispersion and Al2O3support,promotion of water-gas shift reaction,and enhancement of CO oxidation[4,6-9].
For application as an oxygen storage material in TWCs,high surface area is preferred.However,pure CeO2is thermally unstable.For instance,after calcination at 850℃,the surface area of CeO2sample is only 5 m2·g-1[10].The loss of surface area will cause the deterioration ofoxygen storage and release properties.Therefore,thermal stability is one of the most important properties.
In attempt to get CeO2-based materials with high thermal resistance,numerous approaches,including precipitation method[11-12],hydrothermal route[13-15],solgel techniques[16-18],surfactant-assisted approach[19-21]and combustion synthesis[22-23],have been developed to produce CeO2material.Up to now,the most convenientmethod is precipitation,wherein the precursors of cerium mainly include Ceバ and Ceビ,while ammonia is conventionally used as the base.In this method,the H2O2-assisted route developed by Woodhead[24]has been investigated extensively.Under basic condition,Ceバ can be oxidized into Ceビ,in the form of Ce(OH)4or CeO2(OH)2,which will be converted into CeO2by the following hydrolysis and thermal decomposition processes[25-26].However,to the best of our knowledge,there have been no open reports on the investigation of this method employing ammonium carbonate as the precipitant.
In this study,we have developed an ammonium carbonate hydrolysis route based on the H2O2-assisted method,aiming at producing CeO2material with higher thermal stability.In particular,the formation mechanism for the precipitate has also been examined.The textural and structural properties of CeO2oxide are influenced by the precursors.In the ammonium carbonate environment,the growth process as well as the agglomeration of CeO2particles is controlled,which enlarges the grain size of CeO2and facilitates the improvement of thermal stability and reduction property[27].
1 Experimental
1.1 Synthesis
1.1.1 Preparation of undigested precipitates
A solution was prepared by dissolving Ce(NO3)3·6H2O in distilled water,then a freshly standardized H2O2solution (30wt%)was added.The molar ratio of Ce(NO3)3∶H2O2was 1∶1.5.An ammonium carbonate solution (25wt%)was added into the salt solution,under stirring,until pH=8.5.The obtained precipitate was filtered,washed with distilled water until constant pH value,and air-dried at room temperature to obtain CeO2-H.For comparison,an ammonia solution(25wt%)was also used to prepare CeO2precursor following the same procedure,and the obtained sample was labeled as CeO2-C.
1.1.2 Preparation of digested precipitates
An ammonium carbonate solution (25wt%)was added into the mixed solution of Ce(NO3)3·6H2O and H2O2(the molar ratio of Ce(NO3)3∶H2O2was 1∶1.5),under stirring until the pH value of 8.5.The reaction vessel was placed in a water bath,and the solution was heated at 80 ℃ for 12 h,under vigorous stirring.The precipitate was filtered,washed with distilled water,and dried at 70℃for 5 h to obtain sample CeO2-H80.In addition,another sample(CeO2-C80)was also made under the same condition except that ammonia was employed as the precipitant.
1.1.3 Calcination of precipitates
TheCeO2-H80andCeO2-C80sampleswere further calcined at different temperatures in the range of 500~900 ℃ for 3 h.The obtained oxides were labeled as CeO2-Ht and CeO2-Ct,respectively,where t represents the calcination temperature.
1.2 Characterizations
The FTIR measurements were performed on a Nicolet 6700 FT spectrometer,working in the range of 400~4 000 cm-1at a resolution of 4 cm-1using KBr pellet.
The precipitate was analyzed by Raman spectroscopy using a LabRAM HR800 with confocal microscope system.The specimen was illuminated through a 50×objective with 633 nm excitation from a diode laser source at a laser power of 150 mW and with a spot size of 3 μm.Raman spectra were collected in the range of 200~2 000 cm-1.
X-ray photoelectron spectra were obtained on a XSAM 800 spectrometer(KRATOS Corp.),employing Mg Kα radiation (hν=1 253.6 eV)and working at 13 kV and 20 mA.The C1s peak(284.6 eV)was used to calibrate the binding energy.
The weight loss and thermal behavior of the sample were examined by a HCT-2 analyzer(Beijing Science Apparatus Factory,Beijing,China)in flowing nitrogen atmosphere.The sample was heated to 600℃ at a heating rate of 10 ℃·min-1.
The textural properties were determined by N2adsorption-desorption method on a Quantachrome SI instrument.The specific surface area and pore size distribution of the sample were obtained according to Brunauer-Emmett-Teller (BET)method and Barret-Joyner-Halenda (BJH)equation,respectively.Prior to each measurement,the sample was degassed at 300℃for 3 h under vacuum.
X-ray diffraction patterns were recorded using a D/max-rA diffractometer(RIGAKU Corp.)with Cu Kα radiation(40 kV,25 mA,λ=0.154 18 nm).The crystal size was calculated by Scherrer′s equation using the data of(111)peak.
Temperature-programmed reduction (TPR)was determined in a conventional reactor equipped with a thermal conductivity detector.Prior to the analysis,the sample(100 mg)was cleaned by flowing N2at 450℃ for 45 min.The measurement was performed from room temperature to 900℃in a flow of 5%H2/N2(V/V)(20 mL·min-1)at a heating rate of 10 ℃·min-1.
2 Results and discussion
2.1 Formation processes of the precipitates
Composition of the precipitate depends on the kind of cations and anions in the solution.In the present work,Ce3+is employed as cerium precursor,while OH-or CO32-is used as the precipitant,in the presence of hydrogen peroxide[28].
When ammonia is utilized as the precipitant,the following equations are expected during the precipitation and digestion processes[25]:

The proposed mechanism of ammonium carbonate hydrolysis route is as follows[25-26,29]:
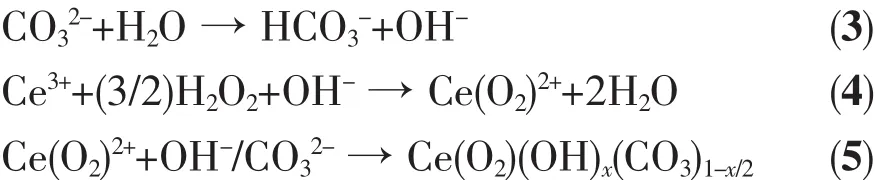
During the heating process of the solution at 80℃,CeO2is gradually formed via:

The dehydration behavior may take place during the whole operation process because the O22-and OH-groups are not stable[25,30].
Fig.1 shows the FTIR spectra of the precipitates before and after heat treatment at 80℃.The intense and broad bands in the 3 000~3 750 cm-1region are normally ascribed to the stretching of surface hydroxyl groups or molecularly chemisorbed H2O[28,31-32].Obviously,three distinguished signals are observed,indicating the coexistence of mono-coordinated, bicoordinated and tri-coordinated hydroxyl groups[28,31-32].The relative intensity of this region for CeO2-H becomes stronger after digestion,whereas CeO2-C shows a different trend.This reveals that the carbonate species is hydrolyzed into hydroxylgroups during the digestion process.The minor band at 2 300~2 400 cm-1corresponds to the coordinated CO2adsorbed at the surface,while the feature at~1 620 cm-1is assigned to the adsorption of H2O[28,33].Several adsorption bands around 1 620,1 380,1 136,1 030 and 913 cm-1are associated with the carbonate species[32,34-35].Among these bands,the one around 1380 cm-1is probably assigned to the carbonate species from the ammonium carbonate precipitator because it can only be observed in CeO2-H,whilst the others are likely caused by atmospheric dioxide[36].

Fig.1 FTIR spectra of precipitates
Raman spectra of the precipitates are presented in Fig.2.It has been reported that bulk CeO2shows a strong peak centered at 456 cm-1or 471 cm-1corresponding to the symmetric breathing mode of the O2-anions[26,37].However,both of the peaks can be found in this work.The broad peak around 608 cm-1is ascribed to the defect site of CeO2crystallite,which seems to be caused by the influence of H2O2[26].The formation of CeO2suggests that the following overall process has occurred[25]:

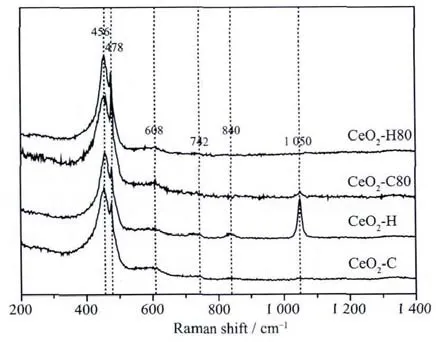
Fig.2 Raman spectra of precipitates
Three bands,locating at about 742,1 050 and 1 350 cm-1,respectively,are likely related to the CO32-species[26].Notably,these spectral features for CeO2-H are the strongest among the samples because of the usage of ammonium carbonate.The reduction of the bands for CeO2-H80 compared to CeO2-H implies that the CO32-species is hydrolyzed during digestion process,in accordance with TG-DTA and IR results.An additional weak band at about 840 cm-1can be observed in the spectra for CeO2-C and CeO2-H,which is indicative of the O-O stretching vibration of η2-peroxide(O22-)species[38],indicating the formation of CeO22+precursors.However,the band vanishes after digestion.This implies that the O22-species is more or less eliminated during the thermal treatment,because the O22--containing species is not stable[25-26].The Raman results confirm the occurrence of Eqs.(3)~(7).
The oxidation states of cerium for the precipitates prepared by different routes are shown in Fig.3.The spectra are very similar,showing six peaks at around 883.0,888.9,898.5,901.1,907.4,and 916.8 eV,respectively.All the spectra are attributed to the diversified states of Ce4+and no signals related to Ce3+are found[39],from which we can conclude that Ceバis fully oxidized into Ceビby H2O2under basic condition.
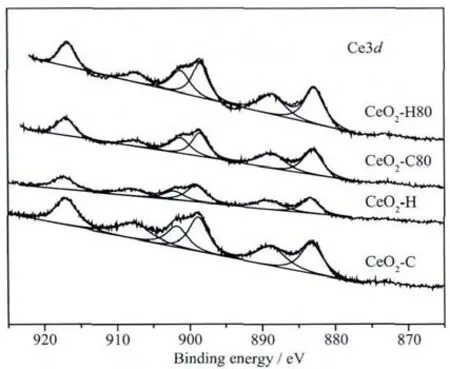
Fig.3 Ce3d XPS spectra for precipitates
The O1s spectra are shown in Fig.4.The peak at about 529.7 eV is characteristic of the O2-anions in bulk CeO2[39],while the peaks at 531.5 and 532.7 eV correspond to OH-and CO32-species,respectively[40-41].It is obvious that the contents of OH-and CO32-species for the digested precipitates are lower than those of the undigested ones,due to the fact that the hydrolysis of CO32-species as well as the dehydration of OH-species takes place during digestion.The relative contents of Ce,O and C are calculated.The relative contents of atoms in CeO2-C are different from CeO2-H.The content of C of CeO2-H is much higher than that of CeO2-C due to the participation of CO32-species during the precipitation procedure.However,the amount of the elements for CeO2-C80 and CeO2-H80 is almost the same,providing another evidence for the occurrence of equation (6)for CeO2-H.It is important to remark that the C element is difficult to evaluate due to the interference of adventitious carbon and physically absorbed CO2[26].
Fig.4 O1s XPS spectra for precipitates
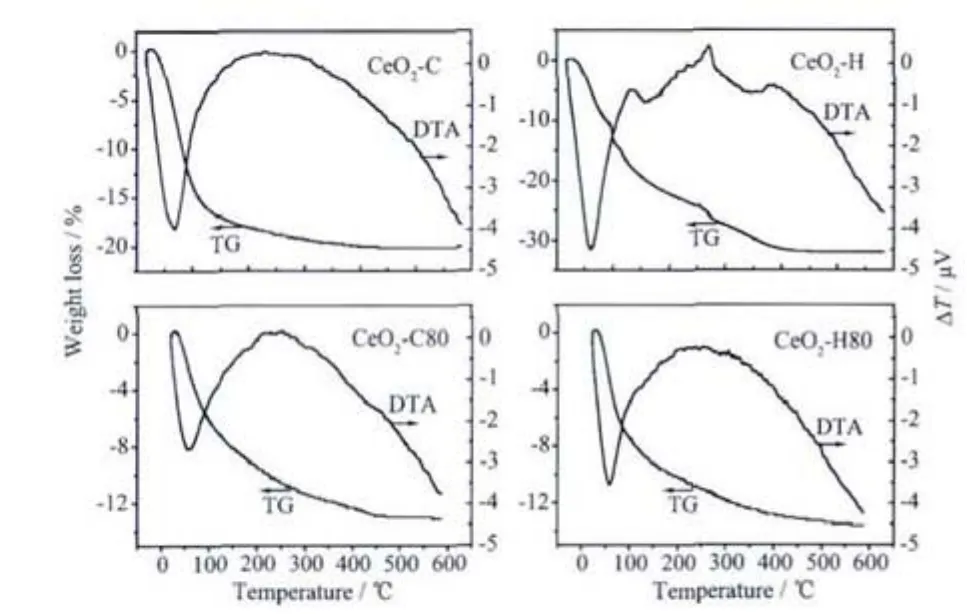
Fig.5 TG/DTA curves of precipitates
Fig.5 displays the TG/DTA curves ofthe precipitates.Both of the precipitates prepared by ammonia show a steady decomposition process,regardless of digestion.For CeO2-C,the total weight loss is about 20.3%,which is much higher than that of CeO2-C80 (13.4%),indicating that the dehydration process,i.e.,equation (2),has occurred during the heat treatment at 80 ℃[30].The DTA curve shows one endothermic peak below 100℃,which is probably ascribed to the elimination ofsurface physicaladsorbed water[30].Since the decomposition process of Ce(O2)(OH)2is a slow and successive process,no obvious exothermal phenomena can be observed.The weight loss procedure of CeO2-H shows complex stages,approximately locating at thetemperature ranges of 25~200 ℃,250~350 ℃ and 350~450 ℃,respectively.The first region appears as a result of the elimination of physically adsorbed water and the decomposition of O22-and hydroxyl groups[25,28,30].The second stageisrelated tothe crystallization of hydroxide particles[30],while the last one originates from the carbonate species[2,28].It seems that the presence of carbonate can affect the decomposition events of hydroxyl species,therefore,several exothermal peaks appear on the DTA curves.After digestion,the overall decomposition behavior of CeO2-H80 is similar to that of CeO2-C80,with a total weight loss of about 13%and an endothermic peak at around 60℃.This implies that the carbonate species has been hydrolyzed into hydroxyl species during the digestion.
2.2 Textural properties of CeO2samples
The surface area of the CeO2samples calcined at different temperatures is illustrated in Fig.6.As expected,the surface area decreases athigher temperatures,regardless of synthetic approaches[19].However,the values are still appreciable.The surface area of CeO2-Ht is superior to that of CeO2-Ct.After calcination at 900℃,the surface area of the CeO2-Ht sample can be 27 m2·g-1,which is rather uncommon for pure CeO2material under such a harsh thermal treatment.This implies that the usage of ammonium carbonate hydrolysis approach allows the formation of CeO2with improved thermal stability.
Fig.7(a)and(b)display the nitrogen adsorption/desorption isotherms and the corresponding pore size distribution curves of the CeO2-C500 and CeO2-H500 samples.According to the IUPAC definition,all the samples feature the isotherm of typeⅣ,typical of a mesoporous material[42].The shape of hysteresis loops indicates the presence of “ink-bottle” and cylindrical pores,which arefavorableforgasand thermal diffusion[42].Actually,the sintering behavior of CeO2-based materials is strongly affected by the pore structure[43].The pore size of CeO2-H500 is larger than that of CeO2-C500,which should be responsible for its improved thermal stability[5,36,44].
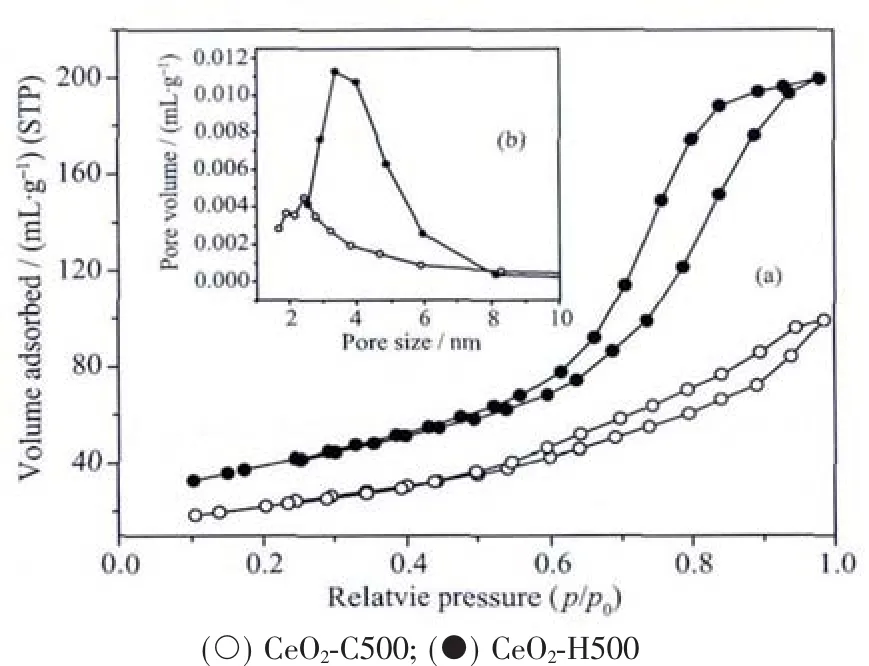
Fig.7 (a)N2adsorption-desorption isotherms of samples prepared by different methods;(b)Corresponding BJH pore size distribution curves
XRD patterns of the precipitates are shown in Fig.8.All the patterns show the diffraction peaks of CeO2with fluorite structure,regardless of preparation routes.The as-prepared samples,i.e.,CeO2-H and CeO2-C,are also characterized by CeO2,indicating that the hydrolysis event of carbonate species as well as the dehydration of hydroxyl groups has occurred before the heat treatment.Crystallite size of the precipitates is calculated using the (111)plane.The grain sizes of CeO2-C(1.9 nm)and CeO2-C80(2.0 nm)are much smaller than those of CeO2-H (3.9 nm)and CeO2-H80 (4.0 nm).This suggests that the crystalline dimension depends on the synthetic routes.As for precipitation process,with progressively increasing concentration of the base solution,the mean magnitude of the individual crystal grain will decrease[45].In the ammonia environment,The OH-species in high concentration attacks Ceビdirectly,resulting in a rapid nucleation rate.Consequently,uniformly small particles are obtained.However,in the case of ammonium carbonate,polymer of carbonate-containing precipitate,i.e.,Ce(O2)(OH)x(CO3)1-x/2,can serve as a precursor of the CeO2oxide.The crystallization of CeO2proceeds through gradual hydrolysis of CO32-,which inhibits the rate of precipitation and enlarges the crystallite size.However,after calcination at 500oC,the crystallite size of CeO2-C500 (7.5 nm)becomes larger than that of CeO2-H500 (4.2 nm).According to the literature[27],the primary particle of CeO2-H is larger than that of CeO2-C,which facilitates the formation of packing “ring”with larger pore space.Generally,the movementofcoarsened particlesofCeO2-Htis expected to be inhibited[44].Moreover,the enlarged pores in CeO2-Ht sinter with more difficulty as longer migration distance is needed for the matter to fill the pores[36].Thus,the CeO2material prepared by ammonium carbonate-hydrolysis method is more thermally stable.

Fig.8 XRD parttens collected from precipitates
2.3 Reduction behavior of CeO2samples
A crucial requirement of CeO2material,especially when used as oxygen storage components for the purification of exhaust gases,is the reduction ability.Thus,the samples were calcined at 500℃and 900℃,and then were employed to characterize the reduction behavior.From Fig.9,the reduction profiles of CeO2-C500 and CeO2-H500 consist of two peaks,which are ascribed to the reduction of surface and bulk oxygen species,respectively[46].Although the outsets of the reduction peaks of the samples are almost the same,the integrated peak area of CeO2-H500 is apparently larger than that of CeO2-C500.This indicates that CeO2-H500 is more reducible and active than CeO2-C500[47].The difference becomes more apparent after calcination at 900℃,especially in the low-temperature region.
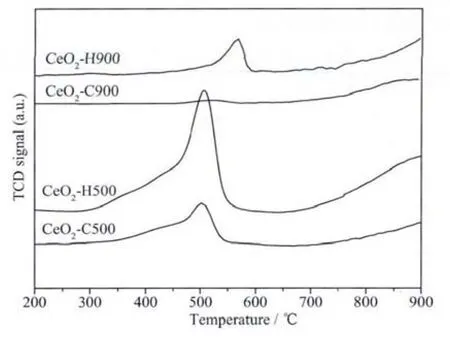
Fig.9 TPR profiles of CeO2samples
3 Conclusions
In this work,CeO2oxide was prepared by a hydrothermal hydrolysis route using ammonium carbonate as the precipitant and hydrogen peroxide as the oxidizer.The title method was compared with the conventionalmethod employing ammonia asthe reactant.The formation mechanism of the precipitate wasalso studied.Afterdigestion,the chemical compositions of the precipitates prepared by the two methods were almostthe same.However,the crystallite size of the precipitates differs from each other greatly.The pore size of the as-prepared CeO2by the hydrolysis procedure is much larger,which facilitates the formation of CeO2oxide with improved texturaland reduction properties.In particular,after calcination at 900℃for3h,theCeO2powder from HA route still remains a surface area of 27 m2·g-1.
[1]Matatov-Meytal Y I,Sheintuch M.Ind.Eng.Chem.Res.,1998,37(2):309-326
[2]Tikhomirov K,Krocher O,Elsener M,et al.A.Appl.Catal.B,2006,64(1/2):72-78
[3]Sahibzada M,Steele B C H,Zheng K,et al.Catal.Today,1997,38(4):459-466
[4]Kašpar J,Fornasiero P,Graziani M.Catal.Today,1999,50(2):285-298
[5]Di Monte R,Kašpar J,Catal.Today,2005,100:27-35
[6]Di Monte R,Fornasiero P,Kašpar J,et al.Appl.Catal.B:Environ.,2000,24:157-167
[7]Kenevey K,Valdivieso F,Soustelle M,et al.Appl.Catal.B:Environ.,2001,29:93-101
[8]Bueno-Lopez A,Such-Basanez I,de Lecea C S M.J.Catal.,2006,244(1):102-112
[9]Nagai Y,Hirabayashi T,Dohmae K,et al.J.Catal.,2006,242(1):103-109
[10]Perrichon V,Laachir A,Abouarnadasse S,et al.Appl.Catal.A,1995,129:69-82
[11]Hernández W Y,Laguna O H,Centeno M A,et al.J.Solid State Chem.,2011,184:3014-3020
[12]Karakoti A S,Kuchibhatla S V N T,Babu K S,et al.J.Phys.Chem.C,2007,111(46):17232-17240
[13]Ahniyaz A,Watanabe T,Yoshimura M.J.Phys.Chem.B,2005,109(13):6136-6139
[14]Si R,Zhang Y W,Wang L M,et al.J.Phys.Chem.C,2007,111(2):787-794
[15]Xian C N,Li H,Chen L Q,et al.Micropor.Mesopor.Mat.,2011,142:202-207
[16]Thammachart M,Meeyoo V,Risksomboon T,et al.Catal.Today,2001,68(1-3):53-61
[17]Fan J,Wu X D,Yang L,et al.Catal.Today,2007,126(3/4):303-312
[18]Ni C Y,Li X Z,Chen Z G,et al.Micropor.Mesopor.Mater.,2008,115:247-252
[19]Terribile D,Trovarelli A,de Leitenburg C,et al.Chem.Mater.,1997,9(12):2676-2678
[20]Terribile D,Trovarelli A,Llorca J,et al.J.Catal.,1998,178(1):299-308
[21]Chen H R,Ye Z Q,Cui X Z,et al.Micropor.Mesopor.Mater.,2011,143:368-374
[22]Mokkelbost T,Kaus I,Grande T,et al.Chem.Mater.,2004,16(25):5489-5494
[23]Heo I,Choung J W,Kim P S,et al.Appl.Catal.B,2009,92(1/2):114-125
[24]Woodhead J L,US Patent,4231893.1980-11-04
[25]Scholes F H,Soste C,Hughes A E,et al.Appl.Surf.Sci.,2006,253(4):1770-1780
[26]Scholes F H,Hughes A E,Hardin S G,et al.Chem.Mater.,2007,19(9):2321-2328
[27]Chen P L,Chen I W.J.Am.Ceram.Soc.,1997,80(3):637-645
[28]Rebellato J,Natile M M,Glisenti A.Appl.Catal.A,2008,339(2):108-120
[29]Li J G,Ikegami T,Mori T,et al.Chem.Mater.,2001,13(9):2913-2920
[30]Djuricˇic' B,Pickering S.J.Eur.Ceram.Soc.,1999,19(11):1925-1934
[31]Binet C,Daturi M,Lavalley J C.Catal.Today,1999,50(2):207-225
[32]Natile M M,Boccaletti G,Glisenti A.Chem.Mater.,2005,17(25):6272-6286
[33]Lin W Y,Frei H.J.Am.Chem.Soc.,2002,124(31):9292-9298
[34]Klissurski D G,Uzunova E L.Chem.Mater.,1991,3(6):1060-1063
[35]Jobbagy M,Marino F,Schobrod B,et al.Chem.Mater.,2006,18(7):1945-1950
[36]Kašpar J,Fornasiero P.J.Solid State Chem.,2003,171(1/2):19-29
[37]Weng X L,Perston B,Wang X Z,et al.Appl.Catal.B:Environ.,2009,90:405-415
[38]Pushkarev V V,Kovalchuk V I,d′Itri J L.J.Phys.Chem.B,2004,108:5341-5348
[39]Zhang G J,Shen Z R,Liu M,et al.J.Phys.Chem.B,2006,110(51):25782-25790
[40]Alifanti M,Baps B,Blangenois N,et al.Chem.Mater.,2003,15(2):395-403
[41]Darnyanova S,Pawelec B,Arishtirova K,et al.Appl.Catal.A,2008,337(1):86-96
[42]Wang J,Wen J,Shen M Q.J.Phys.Chem.C,2008,112(13):5113-5122
[43]Rohart E,Larcher O,Deutsch S,et al.Top Catal,2004,30-31:417-423
[44]Kašpar J,Fornasiero P,Hickey N.Catal.Today,2003,77:419-449
[45]Von Weimarn P P.Chem.Rev.,1925,2(2):217-242
[46]Bruce L A,Hoang M,Hughes A E,et al.Appl.Catal.A,1996,134(2):351-362
[47]Masui T,Peng Y M,Machida K,et al.Chem.Mater.,1998,10(12):4005-4009
猜你喜欢
黑龙江大学自然科学学报(2021年4期)2021-11-19 07:04:56
中小学校长(2021年9期)2021-10-14 14:36:16
环境科技(2017年6期)2018-01-17 08:58:33
中国眼镜科技杂志(2016年9期)2016-12-01 06:37:33
长江蔬菜(2016年10期)2016-12-01 03:05:30
中国科技信息(2016年10期)2016-09-03 03:07:04
当代化工研究(2016年2期)2016-03-20 16:21:18
生物骨科材料与临床研究(2016年6期)2016-03-06 08:20:12
电子器件(2015年5期)2015-12-29 08:42:07
纺织科技进展(2015年1期)2015-11-28 05:56:20
