
堇青石负载Pd和过渡金属氧化物催化剂的表征及其对CO的催化氧化活性
2013-10-12 03:00严小康王志良吴海锁陈英文沈树宝
化工环保 2013年2期
严小康,王志良,吴海锁,黄 琼,陈英文,沈树宝
(1. 南京工业大学 生物与制药工程学院,江苏 南京 210009;2. 江苏省环境科学研究院,江苏 南京 210036)
CO是大气中的主要污染物之一。石油化工企业的生产、化石燃料的燃烧以及机动车的使用均造成大量的CO 排放,导致环境污染日益严重[1-2],CO的消除逐渐成为国内外研究的热点。目前CO的消除方法主要以催化氧化法为主,该法因具有起燃温度低、能耗低、效率高、无二次污染、适用范围广等优点[3],已成为当前工业废气治理领域研究、应用的主流和发展方向,而制备高效、廉价的催化剂是催化氧化法的核心。现有的催化氧化CO催化剂分为贵金属催化剂和非贵金属催化剂,其中贵金属主要包括Pt[4],Pd[5-6],Au[7-8]等;非贵金属主要以过渡元素氧化物的混合、复合物为主,如MnOx,FeOx,CoOx,NiOx,CuO等[9-12]。由于贵金属资源缺乏、价格昂贵,近年来,低含量的贵金属催化剂和高效、廉价的过渡金属氧化物催化剂成为研究热点。目前,在催化氧化CO技术中以Pt、Pd催化剂的应用最为广泛,而Mn的金属氧化物催化剂对CO虽具有较高的催化氧化活性,但在中低温下的催化效果并不理想[13-15]。对于将少量贵金属与过渡金属氧化物结合,制备催化氧化CO催化剂的研究还鲜有报道[16-17]。
本工作以堇青石蜂窝陶瓷(CC)为载体,采用浸渍法制备了CC负载Pd和过渡金属混合氧化物催化剂,记作Pd-M-Mn(M=Cu,Fe,Co,Ni)/CC,考察了过渡金属混合氧化物的组成和Pd负载量对催化氧化CO性能及其稳定性的影响,并对催化剂进行了程序升温还原(H2-TPR)、SEM和XRD表征,探讨了该类催化剂的催化活性与材料理化性能之间的关系。
1 实验部分
1.1 材料、试剂和仪器
CC:12~16目,宜兴非金属化工机械厂有限公司。实验所用试剂均为分析纯。
JSM25900型SEM:日本JEOL电子公司;AXS D8型XRD仪:美国热电公司;CHEMBET-3000型程序升温脱附仪:Quantachrome公司;ASAP-2020MV 3.00H型比表面积和微孔分析仪:美国Micromeritics公司;GC2014型气相色谱仪:日本岛津公司。
1.2 催化剂的制备
将CC浸入质量分数为10%的HNO3溶液中煮沸15 min,采用去离子水多次超声波清洗,以去除CC表面残留的酸,烘干备用。
将一定量的Mn(NO3)2和一定量的M(NO3)x配制成水溶液,称取4 g预处理后的CC,在上述溶液中浸渍6 h,过滤,自然晾干后置于烘箱中80 ℃烘干2 h,500 ℃焙烧5 h,制得Cu-Mn,Co-Mn,Fe-Mn,Ni-Mn混合氧化物负载量均为10%(质量分数,下同)、且Cu,Fe,Co,Ni与Mn的摩尔比均为1∶1的催化剂。
配制一定浓度的Pd(NO3)2浸渍液,采用等体积浸渍法将CC和上述已负载过渡金属混合氧化物的催化剂分别浸渍于上述溶液中6 h,自然晾干后置于烘箱中80 ℃烘干2 h,500 ℃焙烧3 h,制得Pd/CC和Pd-M-Mn/CC催化剂。
1.3 催化剂的活性评价
催化剂催化氧化CO活性评价实验在连续流微型固定床石英反应器中进行,反应器内径为10 mm。称取2.7 g催化剂装入反应器中,然后通入CO与空气的混合气,空速为 10 000 h-1,混合气体组成(体积分数)CO 1.5%,N276.8%,O221.7%。采用程序升温将反应温度从150 ℃升至450 ℃。采用色谱仪在线测定反应气体中CO的含量,计算CO转化率。CO转化率达到100%时的床层温度为T100(℃)。
1.4 催化剂的表征
采用SEM观察催化剂试样的表面形貌;采用XRD仪分析催化剂的晶相, CuKα靶,入射光波长0.154 nm,管电压40 kV,管电流40 mA,扫描范围10°~80°,扫描速率10(°)/min,扫描步长0.05°。采用比表面积和微孔分析仪测定催化剂的比表面积、孔体积及孔径;采用程序升温脱附仪进行H2-TPR实验,热导检测,0.05 g催化剂试样在 200 ℃、N2(流量15 mL /min)气氛中处理1 h ,降温至 100 ℃,在H2(体积分数为5%)与Ar的混合气(流量20 mL/min)中程序升温至700 ℃,升温速率为 10 ℃/min。
2 结果与讨论
2.1 不同的过渡金属混合氧化物对CO转化率的影响
在Pd负载量为0.10%的条件下,不同的过渡金属混合氧化物对CO转化率的影响见图1。由图1可见:CC在反应温度低于250 ℃时,几乎没有催化氧化CO的活性,当反应温度为450 ℃时,CO转化率还不到70%;Pd/CC催化剂催化氧化CO的活性明显提高,在350 ℃时CO转化率达到100%;Pd-MMn/CC催化剂催化氧化CO的活性比Pd/CC催化剂又有明显提高,其中,Pd-Co-Mn/CC催化剂的催化活性最高,在150 ℃时CO转化率为60%,250 ℃时CO转化率达到100%。与Pd/CC催化剂相比,虽然Pd-Cu-Mn/CC、Pd-Fe-Mn/CC和Pd-Ni-Mn/CC催化剂的T100仍需350 ℃,但在200~300 ℃,CO的转化率亦有较大幅度的提高。
CC和M-Mn/CC催化剂的物性参数见表1。由表1可见:CC的比表面积仅为0.6 m2/g;M-Mn/CC催化剂的比表面积均有较大幅度的提高,Fe-Mn/CC催化剂的比表面积最高,达8.0 m2/g,所以在负载过渡金属氧化物之后再负载Pd,可有效提高其分散度,从而提高催化剂的催化活性。虽然Fe-Mn/CC与Co-Mn/CC具有相近的比表面积,但在负载Pd后Pd-Co-Mn/CC催化剂却比Pd/Fe-Mn/CC催化剂表现出更佳的催化活性,由此表明,不同过渡金属氧化物与Pd之间的协同效应也是影响催化剂催化活性的重要因素。
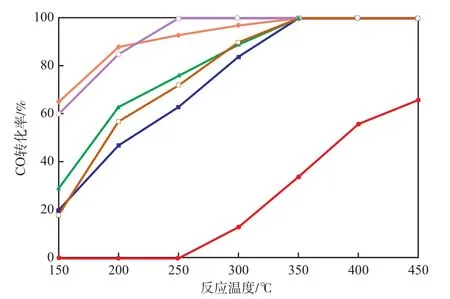
图1 不同的过渡金属混合氧化物对CO转化率的影响

表1 CC和M-Mn/CC催化剂的物性参数
Pd-M-Mn/CC催化剂(Pd负载量为0.10%)的H2-TPR谱图见图2。由图2可见:由于Pd的负载量过低,因而H2-TPR谱图上均未出现PdO的还原峰;Pd-Cu-Mn/CC,Pd-Ni-Mn/CC,Pd-Fe-Mn/CC,Pd-Co-Mn/CC催化剂的最强还原峰依次出现在349,352,550,345 ℃,显现出不同的还原特征峰,表明各催化剂具有不同的还原性能,其中,Pd-Co-Mn/CC催化剂的最强还原峰出现时的温度最低,表明其在较低温度下比Pd-Fe-Mn/CC,Pd-Cu-Mn/CC,Pd-Ni-Mn/CC具有更高的催化活性;Pd-Co-Mn/CC催化剂的扁平状还原峰表明其活性组分在催化剂表面分布较为均匀,且活性物种颗粒较小,与显现尖锐还原峰的Pd-Cu-Mn/CC催化剂相比,Pd-Co-Mn/CC 催化剂具有连续的催化氧化能力,催化剂稳定性更佳。

图2 Pd-M-Mn/CC催化剂的H2-TPR谱图
2.2 Pd-Co-Mn/CC催化剂Pd负载量对CO转化率的影响
Pd-Co-Mn/CC催化剂Pd负载量对CO转化率的影响见图3。由图3可见:随着Pd负载量的增加,CO转化率逐渐增加;未负载Pd时,Co-Mn/CC催化剂在反应温度为200 ℃时的CO转化率仅为15%,450 ℃时CO转化率达到90%;Pd-Co-Mn/CC催化剂(Pd负载量0.01%)150 ℃时的CO转化率不足10%,350 ℃时CO转化率达到100%;Pd-Co-Mn/CC催化剂(Pd负载量0.10%)150 ℃时的CO转化率即可达到87%,200 ℃时CO转化率达到100%;Pd-Co-Mn/CC催化剂(Pd负载量1.00%)在150 ℃时CO转化率即可达到98%,200 ℃时CO转化率达到100%。
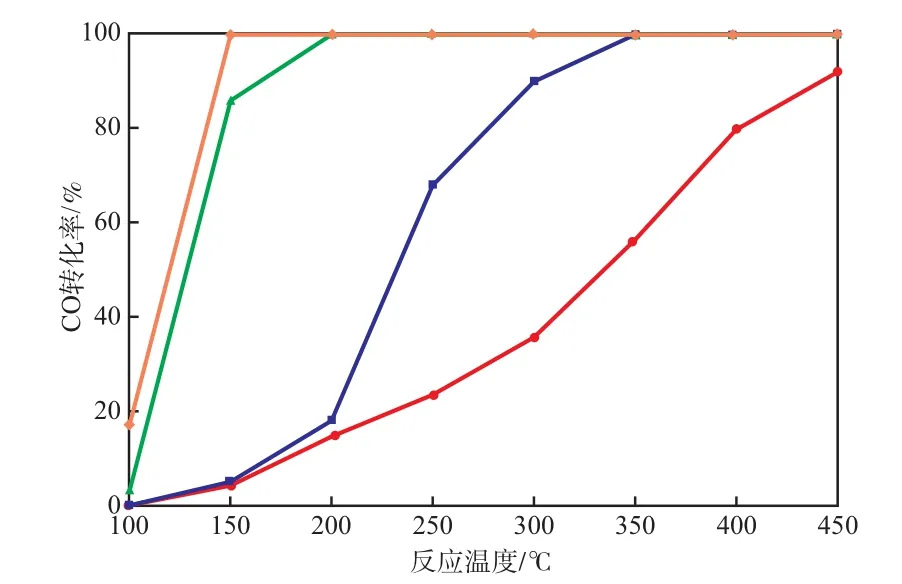
图3 Pd-Co-Mn/CC催化剂Pd负载量对CO转化率的影响
Pd负载前后催化剂的SEM照片见图4。由图4a可见,CC经HNO3溶液处理后结构较为疏松,且具有不同孔径的微孔,有助于活性组分的负载。由图4b可见,未负载Pd 的Co-Mn/CC催化剂的表面出现大量微型颗粒物,整体呈现珊瑚状,层状结构明显。由图4c和图4d可见,Pd-Co-Mn/CC催化剂表面出现了分布较为均匀的不规则长方体针状物,且与载体结合紧密,且图4d中的长方体针状物较图4c中更加密集,表明随着Pd负载量的增大,活性组分因聚集而晶粒增大,导致活性组分的均匀分布程度下降,使催化剂催化活性无法大幅度提升,这也与催化剂活性测试结果一致。

图4 Pd负载前后催化剂的SEM照片
2.3 Pd-Co-Mn/CC催化剂的催化活性稳定性
在反应温度150 ℃条件下,Pd-Co-Mn/CC催化剂(Pd负载量1.00%)催化氧化CO的稳定性实验结果见图5。由图5可见:反应前30 h内CO转化率小幅度下降;随着反应时间的延长,CO转化率均稳定在90%以上。反应100 h后,催化剂表面颜色发生显著变化,由黑色变为棕褐色。

图5 Pd-Co-Mn/CC催化剂(Pd负载量1.00%)催化氧化CO的稳定性实验结果
Pd-Co-Mn/CC催化剂(Pd负载量1.00%)反应前(a)后(b)的XRD谱图见图6。
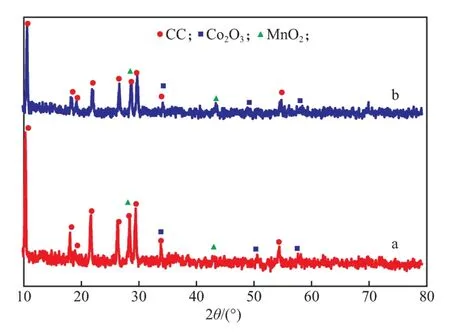
图6 Pd-Co-Mn/CC催化剂(Pd负载量1.00%)反应前(a)后(b)的XRD谱图
由图6可见,反应前后催化剂的XRD谱图中均未出现PdO的特征衍射峰,这可能是因为PdO的衍射峰被CC的部分特征衍射峰所掩盖,或PdO处于高度分散状态而无法被检测到[18]。此外,反应后的催化剂在23°和35°处的衍射峰强度略微变弱,CC、Co2O3和MnO2的特征峰均无显著变化,说明催化剂晶型结构稳定。由此表明,催化剂在反应前30 h催化活性的下降并不是由于活性组分晶型被破坏而造成的,而是由于在催化剂由活性初期逐渐进入活性稳定期的过程中,催化剂表面少量的PdO、Co2O3和MnO2与较高浓度的CO直接接触后立即发生氧化还原反应而生成了类似碳酸盐的物种[19-20],导致部分活性位点的丧失和氧化物中金属价态的下降,因此催化剂在初始反应阶段的催化活性下降,也正是此类氧化还原反应导致了金属氧化物衍射峰强度的减弱和催化剂表面颜色的变化。
3 结论
a)以CC为载体、采用浸渍法制备的Pd-MMn/CC催化剂对CO催化氧化具有良好的催化活性,在Pd负载量为0.10%的条件下,各种过渡金属混合氧化物中Pd-Co-Mn/CC催化剂的催化活性最高,250 ℃时CO转化率达到100%。
b)随着Pd负载量的增加,CO转化率逐渐增加。当Pd负载量为1.00%、反应温度为150 ℃时,CO的转化率达到98%,200 ℃时CO转化率达到100%。
c)在反应温度150 ℃条件下,Pd-Co-Mn/CC催化剂(Pd负载量1.00%)催化氧化CO的反应在前30 h内CO转化率小幅度下降;随着反应时间的延长,CO转化率均稳定在90%以上。反应100 h后,催化剂表面颜色发生显著变化,由黑色变为棕褐色。
[1] 毕玉水,吕功煊. 一氧化碳低温催化氧化研究进展[J]. 分子催化,2003,17(4):314-319.
[2] 吕宏安,冯强. CO低温氧化催化剂研究进展[J]. 广东化工,2008,35 (8):48-49.
[3] Khan F I,Ghoshal A K. Volatile organic compounds control:Best possible techniques[J]. Chem Eng World,1999,34:103-122.
[4] Parthasarathi B,Patil K C,Jayaram V,et al. Ionic dispersion of Pt and Pd on CeO2by combustion method:Effect of metal-ceria interaction on catalytic activities for NO reduction and CO and hydrocarbon oxidation[J]. J Catal,2000,196:293-301.
[5] Guo Yun,Lu Guanzhong,Zhang Zhigang,et al. Effects of ZrO2/Al2O3properties on the catalytic activity of Pd catalysts for methane combustion and CO oxidation[J]. Catal Today,2007,126:441-448.
[6] Marco F,Nicola C,Sergio D R,et al. Pd supported on tetragonal zirconia:Electrosynthesis,characterization and catalytic activity toward CO oxidation and CH4combustion[J]. Appl Catal,B,2005,2005,60:73-82.
[7] Chang Lihsin,Sasirekha Natarajan,Rajesh Baskaran,et al. CO oxidation on ceria and manganese oxidesupported gold catalysts[J]. Sep Purif Technol,2007,58:211-218.
[8] Li Yang,Liao Weiping,Suo Zhanghuai. Influence of KOH modification on TiO2structure and Au/TiO2catalyst activity in CO oxidation[J]. J Fuel Chem Technol,2011,39(1):47-53.
[9] Maryam A,Matin P,Mehdi H. Effect of copper substitution and preparation methods on the LaMnO3catalysis of CO oxidation[J]. Chin J Catal,2010,31:394-403.
[10] Luo Jinyong,Meng Ming,Li Xiang,et al. Mesoporous Co3O4-CeO2and Pd/Co3O4-CeO2catalysts:Synthesis,characterization and mechanistic study[J].J Catal,2008,254:310-324.
[11] Luo Jinyong,Meng Ming,Yao Jinsong,et al. Onestep synthesis of nanostructured Pd-doped mixed oxides MOx-CeO2(M=Mn,Fe,Co,Ni,Cu) for ef cient CO and C3H8total oxidation[J]. Appl Catal,B,2009,87:92-103.
[12] 陈敏,郑小明. Ag-Fe复合氧化物催化剂上CO的氧化性能[J]. 石油化工,2000,29(1):914-916.
[13] 尉士民,杜芳林. CO氧化催化剂研究进展[J]. 工业催化,2003,11(1):32-36.
[14] 杨肖,贾志刚,季生福,等. Al-MnO2/SBA-15催化剂的制备及其催化燃烧甲醛的性能[J]. 化工环保,2011,31(4):369-374.
[15] Shinji Kudo,Taisuke Maki,Masahiro Yamada,et al. A new preparation method of Au/ferric oxide catalyst for low temperature CO oxidation[J]. Chem Eng Sci,2010,65:214-219.
[16] Hu Rongrong,Xie Lanying,Ding Shi,et al. CO oxidation and oxygen-assisted CO adsorption/desorption on Ag/MnOxcatalysts[J]. Catal Today,2008,131:513-519.
[17] Xu Xiuyan,Li Jinjun,Hao Zhengping. CeO2-Co3O4Catalysts for CO Oxidation[J]. J Rare Earths,2010,32(11):43-47.
[18] 李永峰,刘祖超,麦荣坚,等. Pd基无涂层整体式催化剂上甲苯催化燃烧净化研究[J]. 燃料化学学报,2011,39 (9):712-716.
[19] 邹旭华,齐世学,索掌怀,等. CO低温氧化 Au/Al2O3催化剂的失活及稳定性[J]. 催化学报,2006,27(2):161-165.
[20] Kim Moon Hyeon,Kim Dong Woo. Parametric study on the deactivation of supported Co3O4catalyst for low temperature CO oxidation[J]. Chin J Catal,2011,32(5):762-770.
猜你喜欢
化学工程师(2023年1期)2023-02-17
理化检验-化学分册(2020年12期)2021-01-26
陶瓷学报(2020年6期)2021-01-26
上海农业科技(2019年1期)2019-02-22
中学生数理化·中考版(2018年11期)2019-01-31
教学考试(高考化学)(2018年5期)2018-12-06
中国果业信息(2018年5期)2018-01-17
淮南师范学院学报(2015年3期)2015-03-22
河北科技大学学报(2015年5期)2015-03-11
无机化学学报(2014年4期)2014-02-28
