基于曙红Y 氧化性猝灭的高活性和高稳定的可见光催化产氢催化剂
2013-09-17 06:59吕功煊
物理化学学报 2013年8期
李 波 吕功煊
(1中国科学院大学兰州化学物理研究所,羰基合成与选择氧化国家重点实验室,兰州730000;2中国科学院大学,北京100049)
1 引言
光催化分解水生成氢气和氧气是将太阳能转换为清洁、高效化学能的最佳途径,是目前国内外新能源研究领域的研究热点之一.1-13在诸多光催化剂材料中TiO2以其优良的物理化学性能被广泛应用,但TiO2的带隙较宽,只能响应太阳光中含量仅有4%的紫外光.扩展半导体催化剂对太阳光的响应能力是构建高效光催化产氢体系的关键问题.染料敏化作为一种有效提高宽禁带半导体在可见光区响应的手段在染料敏化太阳能电池(DSSCs)领域取得很大成功的同时,在染料敏化半导体光催化制氢领域的研究也备受关注.14-17
研究表明,虽然以荧光素类染料为敏化剂的半导体光催化产氢体系能够表现出高的产氢活性,但是只有吸附在半导体表面的染料才具有敏化效应并且产氢的稳定性难以长期保持.18,19为了提高此类染料敏化半导体光催化产氢的效率和稳定性,人们通过调节染料与半导体的界面键合方式提高敏化体系的吸光效率及电子的传递速率.例如Li等20利用曙红Y(EY)与Fe3+的耦合效应,构建了多层染料敏化的EY-Fe3+-Pt/TiO2光催化剂,Fe3+对染料耦合效应不仅增强了催化剂对可见光的吸收效率,而且Fe3+还作为电子传递的介质增强了不同染料分子层之间的电子传递效率,从而使光催化产氢效率大幅提高.Abe等21利用硅胶偶联剂将曙红Y固载在TiO2表面,显著提高了染料敏化剂的析氢稳定性.虽然这些方法有效地提高了曙红Y在TiO2表面的吸附量以及激发态染料向半导体注入电子的效率,但是曙红Y敏化半导体光催化产氢体系中由于牺牲试剂如三乙醇胺(TEOA)或乙二胺四乙酸(EDTA)等的存在,激发态曙红Y分子易于发生还原性猝灭生成不稳定的还原态中间体,导致染料的降解,从而使光催化产氢效率和稳定性降低.22因此如何抑制易降解还原态染料物种的生成是提高染料敏化体系光催化产氢活性的关键.Grätzel等23以甲基紫精(MV2+)作为电子传递剂构建了具有较好活性的均相光催化产氢体系,并证明H2是由于 MV2+还原态(MV+•)在 PtO2表面将水还原产生的.在荧光素类染料的光化学性能的研究中,Islam等24研究发现在过量TEOA的存在条件下,通过可见光激发低浓度的EY能将MV2+还原为稳定存在的MV+•阳离子,并且这一过程中激发态EY主要发生氧化性猝灭,抑制了不稳定中间体的生成,这为构建高效、稳定的基于荧光素染料为敏化剂的光催化产氢体系提供了有利条件.但是迄今为止,这方面的研究较少.
本文将电子传递剂MV2+引入染料敏化体系中,以EY为敏化剂,TiO2为半导体光催化剂、Pt为产氢助催化剂,TEOA为电子给体,构建了一类高效、稳定的可见光催化产氢体系.通过对光催化体系产氢活性、UV-Vis吸收光谱、荧光光谱以及光电化学性能测试,深入讨论了MV2+对曙红Y敏化Pt/TiO2光催化体系活性和稳定性的促进作用,并结合前人的研究结果提出了MV2+参与的曙红Y敏化Pt/TiO2光催化体系反应机理.
2 实验部分
2.1 试剂与仪器
EY、三乙醇胺(分析纯,国药集团化学试剂有限公司)、甲基紫精(纯度98%,百灵威科技有限公司)、氯铂酸(分析纯,沈阳市科达试剂厂),使用前未经进一步纯化处理.以P25二氧化钛(德国Degussa公司,比表面积50 m2·g-1,70%锐钛矿,粒径~30 nm)为光催化剂.紫外-可见吸收光谱采用HP-8453型紫外可见吸收光谱仪进行测定(美国惠普公司);稳态荧光光谱采用FluoroMax-4型荧光光谱仪(法国Horiba Scientific公司)测定.
2.2 Pt担载TiO2(Pt/TiO2)的制备25
Pt/TiO2通过氢气热还原法制备,具体步骤如下:搅拌条件下将1.0 g P25粉末和一定量(1.0%(w)Pt)的H2PtCl6·6H2O水溶液混合,用红外灯将溶液蒸干,将产物在400°C空气气氛下煅烧1 h,然后在400°C氢气气氛下还原1 h,得到Pt/TiO2.
2.3 光催化产氢反应
光催化反应在一个容积为155 mL的侧面带有平面窗口(有效光照面积约为10 cm2)的石英反应瓶中进行的,瓶口用硅橡胶垫密封,以便定时取样分析.将Pt/TiO2光催化剂分散于80 mL 15%(体积分数,φ)TEOA水溶液中,然后加入一定量的染料,混合均匀.光照反应开始前,混合体系超声分散3 min,然后用高纯氩气吹扫置换40 min,排除反应体系中的氧气.反应过程中利用电磁搅拌,使催化剂一直处于悬浮状态.以300 W金属卤钨灯作为光源,并配有420 nm截止滤光片滤去紫外光.反应气相产物中的氢气含量用气相色谱(Agilent 6820)分析,检测器为热导检测器(TCD),载气为氩气,13X分子筛填充柱,外标法定量.
2.4 光电性质测试
工作电极是通过滴涂法制备的.首先将100 mg Pt/TiO2粉末催化剂加入到10 mL蒸馏水中经过超声波分散10 min,得悬浮液备用.用微量移液器移取250 μL悬浮液滴到已经处理干净的氧化铟锡(ITO)导电玻璃导电面上并在250 W的红外灯下缓慢烘干作为工作电极.光电化学实验是在一个自行设计制作的三电极电化学池中进行,光电化学池三电极与CHI-660型电化学工作站相连,对电极为铂丝电极,参比电极为饱和甘汞电极(SCE).光源为300 W氙灯,并配有420 nm截止滤光片,支持电解质为15%(φ)TEOA(pH 7)水溶液.工作电极的几何受光面积为1.0 cm2.
3 结果与讨论
3.1 MV2+的存在对曙红敏化Pt/TiO2光催化产氢体系活性和稳定性的提高
在可见光条件下(>420 nm),以TEOA为牺牲试剂时,测试了MV2+的加入对EY敏化Pt/TiO2悬浮光催化体系产氢活性及稳定性的影响,其它条件保持一致,产氢量随时间的变化曲线如图1所示.由图可见,在反应的初始阶段(<4 h),两个体系都能保持相对稳定的产氢活性,但由于MV2+的加入,EY-MV2+-Pt/TiO2体系的产氢活性明显高于EY-Pt/TiO2的,产氢速率由35.3 μmol·h-1提高到72.8 μmol·h-1.另外,随着反应时间的延长,6 h后,EY-Pt/TiO2体系的产氢活性明显降低,8 h之后,氢气产量不再增加;与此相反,EY-MV2+-Pt/TiO2体系在10 h内产氢活性保持稳定(图1a).为了进一步考察MV2+对EY-Pt/TiO2体系的产氢稳定性能的促进作用,本文测试了EYMV2+-Pt/TiO2体系在长时间范围内(60 h)的产氢性能,体系每隔12 h置换一次,结果图1b所示.可以看出,在每一个循环中,EY-MV2+-Pt/TiO2体系的产氢活性较为稳定,在所研究的时间范围内,活性下降较为缓慢,反应4次后,活性与第一次相比有所下降,但不明显,反应60 h,总产氢量达到了4096 μmol,基于染料分子的反应转化数(TON)为1024.而在相同光照条件下,含有MV2+的反应体系中缺少EY或者TEOA均没有产氢活性,因此,排除了MV2+作为光敏剂或者电子牺牲试剂的EY-Pt/TiO2体系产氢活性的可能性.以上实验表明,在EY-Pt/TiO2体系中加入MV2+可明显促进该体系的产氢活性和稳定性能,这可能与MV2+作为良好的电子传递剂,有效提高了体系中激发态EY的利用效率有关.
图2a为经过离心除去Pt/TiO2悬浮颗粒并稀释5倍后EY-Pt/TiO2体系反应前后UV-Vis的吸收光谱.从图中可以看出,反应10 h后该光催化体系中EY在520 nm处光吸收强度明显降低,仅为初值的11%,且光的吸收发生了明显的蓝移,该结果表明EY在反应过程中发生了明显的脱卤作用22,26并发生降解,从而导致了EY-Pt/TiO2体系光催化产氢活性的降低和失活.与之相比,当加入MV2+之后,EYMV2+-Pt/TiO2光催化体系中EY的稳定性较高(图2b),反应48 h后反应体系中EY的浓度仍为初始的76%,说明MV2+的参与可有效地抑制EY的降解,使得该体系在可见光照下保持较高的产氢活性和稳定性.另外,从图2b还可以看出,反应60 h后,EY对可见光的吸收强度快速降低,说明EY发生了强烈的降解,这可能与体系中MV2+在长时间反应过程中的失活有关(反应后期溶液的颜色并没有呈现由于MV+生成而显示的紫色),MV2+丧失了电子中继的能力,该结果与图1b所示的EY-MV2+-Pt/TiO2体系反应56 h后活性开始迅速降低直至失活一致.
3.2 MV2+参与的高效电子传递机制
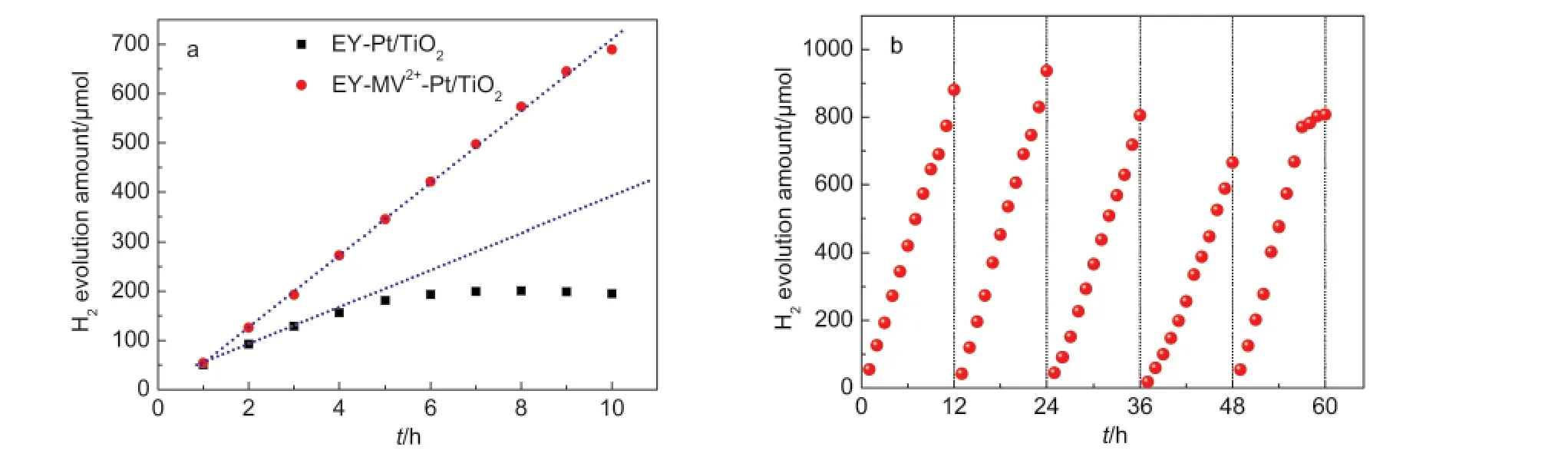
图1 (a)EY-Pt/TiO2和EY-MV2+-Pt/TiO2体系的产氢量随时间的变化曲线,(b)EY-MV2+-Pt/TiO2体系在循环反应中的产氢稳定性能测试Fig.1 (a)Time courses of H2evolution over EY-Pt/TiO2and EY-MV2+-Pt/TiO2systems;(b)stability test for H2evolution over EY-MV2+-Pt/TiO2system in repeated reaction runs
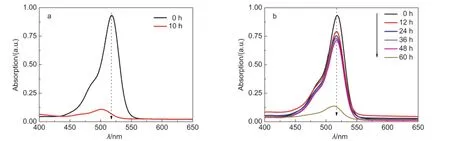
图2 (a)EY-Pt/TiO2和(b)EY-MV2+-Pt/TiO2体系在不同反应时刻的UV-Vis吸收光谱Fig.2 (a)Time-dependent UV-Vis absorption spectra of(a)EY-Pt/TiO2and(b)EY-MV2+-Pt/TiO2systems
Shimidzu等22的研究成果表明,在EY-Pt/TiO2光催化产氢体系中需要加入电子牺牲试剂TEOA才能使之表现出明显的产氢活性,这是因为受光激发形成的3*EY2-极易发生电荷复合,光生电子不能有效地传递到Pt/TiO2表面还原水产氢.当TEOA存在时,3*EY2-能够被TEOA还原,将其转化为具有强还原性的EY3-•自由基,如图3(a)所示.虽然TEOA对3*EY2-的原性猝灭有效地避免了3*EY2-电荷的复合,但生成的中间体EY3-•自由基极不稳定容易发生自身的分解,因而导致EY-Pt/TiO2光催化体系产氢活性的降低和失活.当在EY-Pt/TiO2光催化产氢体系中加入了MV2+之后,将改变以TEOA为电子给体对3*EY2-进行还原性猝灭为主的电子传递机制,Islam等24发现在过量TEOA和MV2+存在的条件下,通过可见光激发低浓度的EY生成的3*EY2-经过氧化性猝灭和还原性猝灭将MV2+化为稳定存在的还原态阳离子自由基MV+•,而且这两种猝灭方式的反应速率常数分别为2.0×1010和2.5×107mol·L-1·s-1,因此,在MV2+参与的条件下3*EY2-主要发生氧化性猝灭,图3(b)给出了TEOA和MV2+存在条件下EY-MV2+-Pt/TiO2光催化体系的电子传递机理.在氧化猝灭中,3*EY2-被MV2+氧化生成EY-•,有效地避免了不稳定中间体EY3-•的生成,光生电子通过较为稳定的MV+•转移至Pt/TiO2表面还原水产氢,EY-•被TEOA还原再生继续吸收入射光.在还原路径中,3*EY2-先被TEOA还原为不稳定中间体EY3-•,而后EY3-•被溶液中的MV2+迅速氧化,避免了EY3-•的积累.因此,由于在EY-Pt/TiO2光催化产氢体系中引入了电子传递剂MV2+,有效避免了不稳定中间体EY3-•的形成与积累,大大降低了EY在光催化反应中的降解速率,提高了体系的产氢稳定性.
此外,图4给出的EY-Pt/TiO2和EY-MV2+-Pt/TiO2光催化产氢体系的稳态荧光光谱结果表明,MV2+的加入并没有对EY的荧光发射性质产生明显的影响,其荧光强度只有略微降低,而且MV2+加入前后EY的荧光寿命分别为1.28和1.27 ns,这表明在MV2+和EY的单重激发态之间直接进行有效的电子传递的概率很低.因此,在EY-MV2+-Pt/TiO2体系中,3*EY2-仍是光催化产氢化反应过程中非常重要的反应中间体,进一步证实了我们对MV2+存在条件下EY-MV2+-Pt/TiO2光催化体系的电子传递机理推测的合理性.
3.3 光电化学特性
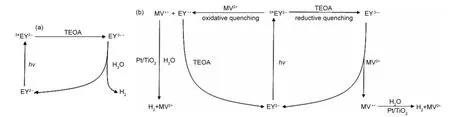
图3 敏化体系(a)EY-Pt/TiO2和(b)EY-MV2+-Pt/TiO2光催化产氢机理Fig.3 Mechanism of photocatalytic hydrogen production of(a)EY-Pt/TiO2and(b)EY-MV2+-Pt/TiO2sensitized systems TEOA:triethanolamine
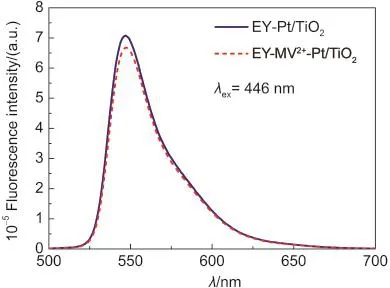
图4 EY-Pt/TiO2和EY-MV2+-Pt/TiO2体系的荧光光谱Fig.4 Fluorescence spectra of EY-Pt/TiO2and EY-MV2+-Pt/TiO2systems
图5 所示的三条曲线为可见光照条件下(λ≥420 nm)同一工作电极(Pt/TiO2薄膜电极)分别在TEOA水溶液、含有EY的TEOA水溶液以及含有EY和MV2+的TEOA水溶液中测得的瞬态光电流曲线.由图5中的曲线a可以看出,在TEOA水溶液中,Pt/TiO2光电极对可见光有一定的响应能力,这是由于Pt纳米颗粒担载的TiO2表面存在等离子体共振效应导致的,27但其光电流密度较小,仅有2.5 μA·cm-2;当溶液中存在光敏剂EY时,Pt/TiO2光电极的光电流密度增加至12 μA·cm-2(曲线b),这是由于EY对Pt/TiO2光电极的敏化作用提高了其对可见光的响应能力.在TEOA水溶液同时加入了光敏剂EY和电子传递剂MV2+后测量得到的瞬态光电流曲线与在上述两种溶液中测得的光电流曲线有很大不同,如图5曲线c所示.在光照过程中曲线c的光电流密度几乎以恒定的增长速率稳步增加,而后增长速度减缓达到平衡状态;当关闭光源后光电流也没有骤然降低,而是以几乎恒定的速率降低.从图3(b)所示电子传递机制可以看出,在TEOA和MV2+存在的条件下,通过光照激发体系中的EY可以将MV2+转化为具有强还原性的MV+•阳离子自由基,而MV+•在电子传递过程中是主要向Pt/TiO2表面注入电子的中间体,与没有MV2+存在的EY敏化Pt/TiO2光催化体系中的还原态中间体EY3-•自由基相比可以更稳定的存在,不会发自身的分解和猝灭.因此,这种特殊的瞬态光电流曲线表明在EY敏化Pt/TiO2光催化产氢体系中,MV2+作为良好的电子传递剂可以更高效地将电子从染料激发态及还原态俘获电子,并将电子储存在更加稳定的MV+•阳离子自由基,一方面提高了体系中EY的循环再生效率,抑制了EY的降解,另一方面,有效提高了光生电子的有效转化效率.
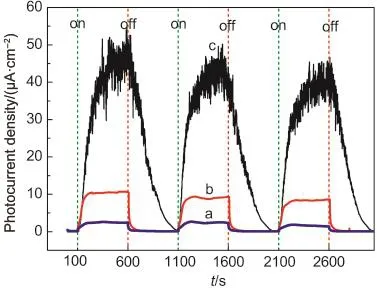
图5 可见光照下Pt/TiO2光电极分别在(a)TEOA水溶液、(b)1×10-4mol·L-1EY的TEOA水溶液和(c)含有1×10-4 mol·L-1EY和1×10-3mol·L-1MV2+的TEOA水溶液测得的瞬态光电流-时间曲线Fig.5 Transient photocurrent-time curves of Pt/TiO2in(a)TEOAaqueous solution,(b)TEOAaqueous solution with 1×10-4mol·L-1EY,and(c)TEOAaqueous solution with 1×10-4mol·L-1EY and 1×10-3mol·L-1MV2+
3.4 EY浓度对敏化体系产氢活性的影响
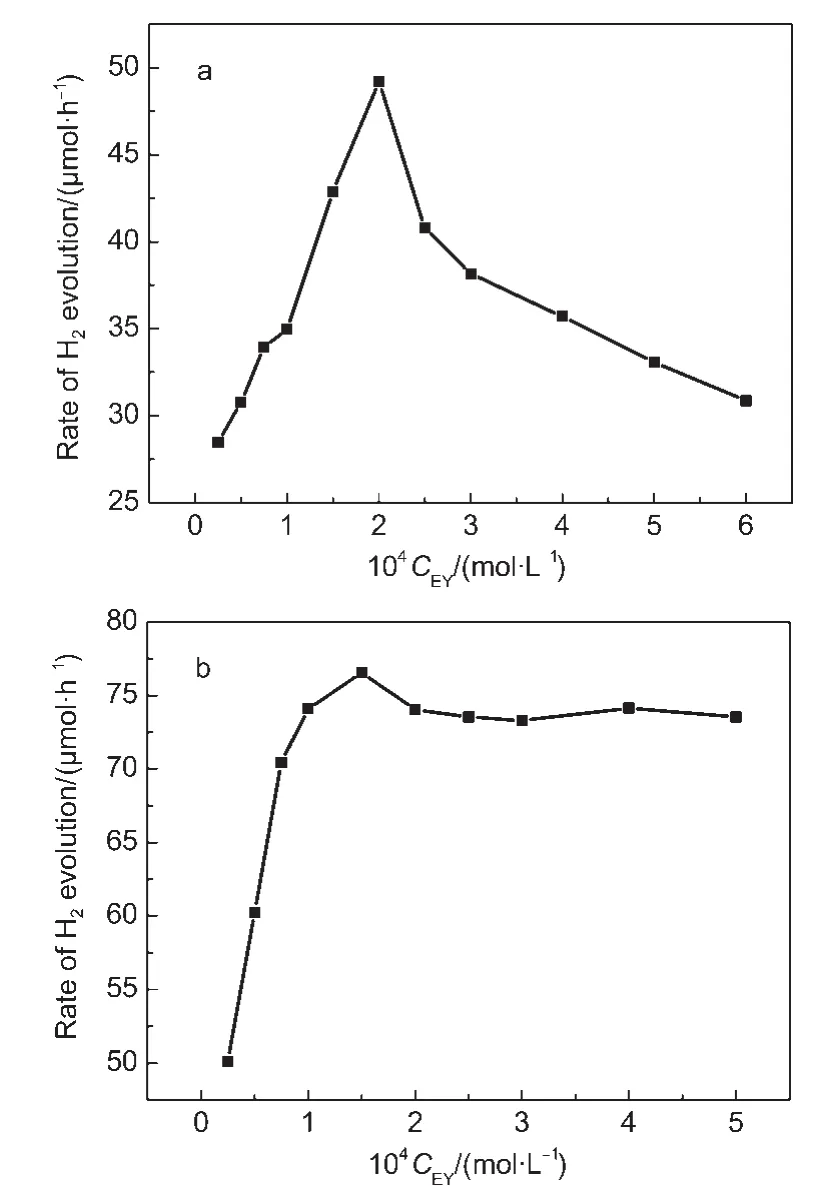
图6 (a)EY-Pt/TiO2和(b)EY-MV2+-Pt/TiO2体系在不同EY浓度条件下产氢量随时间的变化曲线Fig.6 Time courses of H2evolution over(a)EY-Pt/TiO2 and(b)EY-MV2+-Pt/TiO2systems with different concentrations of EY
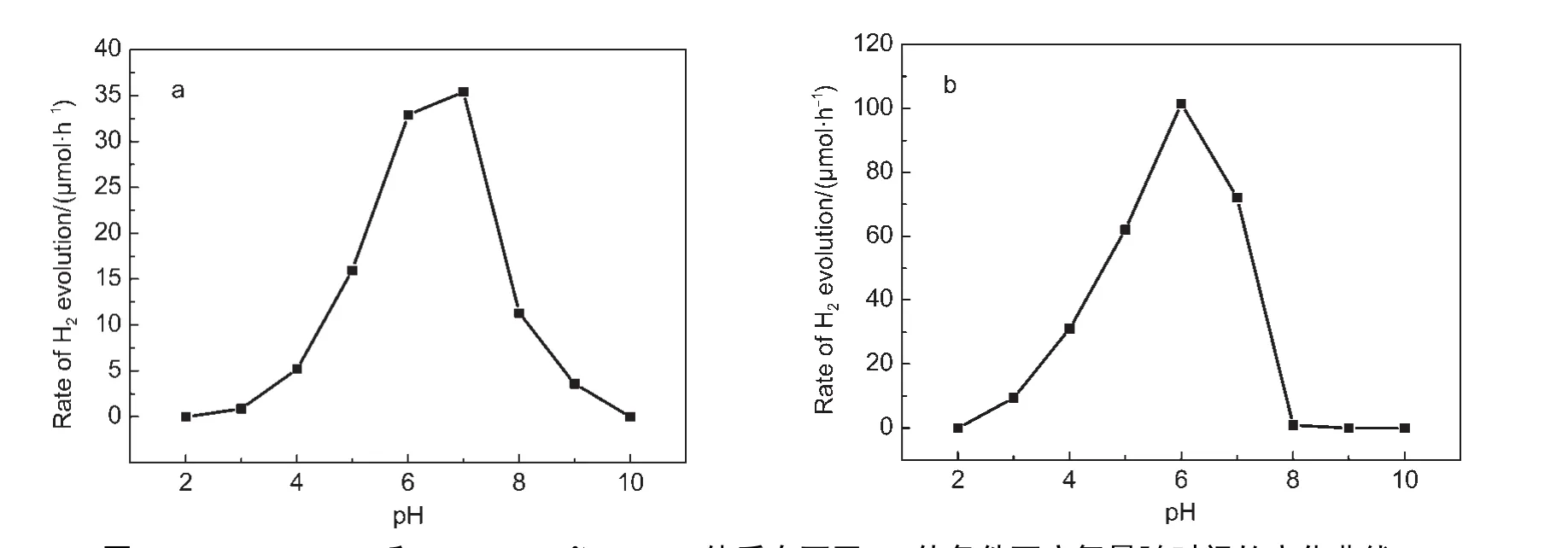
图7 (a)EY-Pt/TiO2和(b)EY-MV2+-Pt/TiO2体系在不同pH值条件下产氢量随时间的变化曲线Fig.7 Time courses of H2evolution over(a)EY-Pt/TiO2and(b)EY-MV2+-Pt/TiO2systems with different pH values
图6 a为EY-Pt/TiO2体系产氢活性随EY浓度变化曲线,EY的浓度较低时(低于2.0×10-4mol·L-1),随着EY浓度的增加,产氢速率随之增加;而当EY的浓度高于2.0×10-4mol·L-1后,产氢活性会随着EY浓度的增加而降低.造成这种现象的原因之一可能是溶液中游离态染料的屏蔽效应.在较低EY浓度下,随着EY浓度的增加,吸附在催化剂表面的染料浓度也会随之增加,直至饱和吸附,催化剂表面染料吸附量的增加有利于提高催化剂表面染料对可见光的吸收效率,以及光生电子向半导体表面的注入,因此在较低浓度下产氢活性会随着EY浓度的增加而升高;然而,当EY浓度过高时,体系中游离态的EY吸收大部分入射光而激发,降低了照射到吸附在催化剂表面染料的光量子数,但是受激发的游离态EY极易发生猝灭,向半导体表面注入电子的效率很低,影响了光催化产氢效率.另一方面浓度猝灭效应也是导致高EY浓度时析氢速率降低的重要原因之一.当染料度过高时,激发态的染料物种与另一个基态或激发态的染料物种碰撞,通过非辐射跃迁、辐射跃迁和系间窜跃导致激发态染料的失活.28
与EY-Pt/TiO2体系不同,在EY-MV2+-Pt/TiO2体系中,较低浓度范围内产氢活性随着EY浓度的增加而增强,但是,当EY浓度大于1.0×10-4mol·L-1后,其产氢活性随EY浓度的增加基本保持不变,如图6b所示.造成两种产氢体系产氢活性随着EY浓度呈现不同变化规律的原因是由于MV2+在电子传递过程中对光生电子有效的弛豫效应.在TEOA存在时,受光激发的EY能够将MV2+还原生成较为稳定存在的MV+•阳离子自由基,而图5给出的瞬态光电流-时间曲线表明,这种稳定MV+•阳离子自由基能够将电子有效地传递到Pt/TiO2表面.因此,MV2+存在条件下EY敏化Pt/TiO2光催化体系受染料的屏蔽效应及浓度猝灭效应较小,这也进一步证明了图3b所示产氢机理是可能的.
3.5 pH值对敏化体系产氢活性的影响
对于光催化制氢反应而言,溶液的pH值对析氢活性有显著的影响,图7给出了EY-Pt/TiO2和EYMV2+-Pt/TiO2体系在不同pH值条件下产氢活性结果.在以TEOA为电子给体的光催化分解水制氢体系中,两种敏化体系的析氢活性的最佳pH值都接近为中性环境,分别为7和6,无论是强酸性还是强碱性都不利于放氢.这是因为溶液的pH值会影响TEOA的存在状态,在强酸性条件下由于三乙醇胺的质子化,使其向EY提供电子的能力减弱,29影响了EY的循环再生.而在强碱性条件下,一方面由于EY的-COO-以及催化剂表面的-OH是以去质子化的形式存在的,由于静电排斥作用,影响了染料在催化剂表面吸附;另一方面,较高pH值下,由于析氢电位升高不利于氢气的产生,故两种染料光催化产氢体系均在近中性溶液条件下显示了较好的活性.
4 结论
通过在EY敏化Pt/TiO2光催化体系中引入电子传递剂MV2+成功构建了高活性与稳定性的可见光催化产氢体系.EY敏化Pt/TiO2光催化体系在MV2+的协助下可以通过氧化性猝灭和还原性猝灭两种电子传递路径将电子从EY传递到Pt/TiO2表面.这种高效的电子传递机制,一方面有效提高了电子从EY向光催化剂Pt/TiO2的传递速率,促进了EY的循环再生,从而提高了对可见光的吸收效率;另一方面有效地抑制了不稳定中间体EY3-•的生成,降低了EY的降解速率,使体系的产氢稳定性得到了明显提高.pH值显著影响两种染料敏化体系的产氢活性,强酸性和强碱性条件均不利于产氢.此外,两种敏化体系的瞬态光电流以EY浓度对产氢活性影响存在明显差异,其原因是由于MV2+作为电子传递剂在光照条件下被还原生成稳定的中间体MV+•,提高了光生电子的传递和利用效率.
(1)Zhou,P.;Lu,G.X.;Ma,J.T.J.Mol.Catal.(China)2011,25(4),328.[周 鹏,吕功煊,马建泰.分子催化,2011,25(4),328.]
(2) Zhou,P.;Zhao,C.J.;Dong,W.P.;Lu,G.X.J.Mol.Catal.(China)2012,26(3),265.[周 鹏,赵成坚,董文平,吕功煊.分子催化,2012,26(3),265.]
(3)Wu,Y.Q.;Lu,G.X.;Li,S.B.J.Mol.Catal.(China)2004,18(2),125.[吴玉琪,吕功煊,李树本.分子催化,2004,18(2),125.]
(4) Elvington,M.;Brown,J.;Arachchige,S.M.;Brewer,K.J.J.Am.Chem.Soc.2007,129(35),10644.doi:10.1021/ja073123t
(5) Li,Q.;Guo,B.;Yu,J.G.;Ran,J.R.;Zhang,B.H.;Yan,H.J.;Gong,J.R.J.Am.Chem.Soc.2011,133(28),10878.doi:10.1021/ja2025454
(6)Wu,Y.Q.;Lu,G.X.J.Mol.Catal.(China)2001,15(6),467.[吴玉琪,吕功煊.分子催化,2001,15(6),467.]
(7)Silva,L.A.;Ryu,S.Y.;Choi,J.;Choi,W.Y.;Hoffmann,M.R.J.Phys.Chem.C 2008,112(32),12069.doi:10.1021/jp8037279
(8)Teets,T.S.;Nocera,D.G.Chem.Commun.2011,47(33),9268.doi:10.1039/c1cc12390d
(9)Wang,X.;Maeda,K.;Thomas,A.;Takanabe,K.;Xin,G.;Carlsson,J.M.;Domen,K.;Antonietti,M.Nat.Mater.2008,8(1),76.
(10)Yan,H.J.;Yang,J.H.;Ma,G.J.;Wu,G.P.;Zong,X.;Lei,Z.B.;Shi,J.Y.;Li,C.J.Catal.2009,266(2),165.doi:10.1016/j.jcat.2009.06.024
(11) Yu,J.;Qi,L.;Jaroniec,M.J.Phys.Chem.C 2010,114(30),13118.doi:10.1021/jp104488b
(12) Zheng,X.H.;Zhang,B.;Li,Q.L.;Jin,Z.S.J.Mol.Catal.(China)1991,5(4),340.[郑新华,张 兵,李庆霖,金振声.分子催化,1991,5(4),340.]
(13) Min,S.X.;Lü,G.X.Acta Phys.-Chim.Sin.2011,27(9),2178.[敏世雄,吕功煊.物理化学学报,2011,27(9),2178.]doi:10.3866/PKU.WHXB20110904
(14)Chen,K.S.;Liu,W.H.;Wang,Y.H.;Lai,C.H.;Chou,P.T.;Lee,G.H.;Chen,K.;Chen,H.Y.;Chi,Y.;Tung,F.C.Adv.Funct.Mater.2007,17(15),2964.
(15)Kubo,W.;Murakoshi,K.;Kitamura,T.;Yoshida,S.;Haruki,M.;Hanabusa,K.;Shirai,H.;Wada,Y.;Yanagida,S.J.Phys.Chem.B 2001,105(51),12809.doi:10.1021/jp012026y
(16) Li,Y.X.;Xie,C.F.;Peng,S.Q.;Lu,G.X.;Li,S.B.J.Mol.Catal.A:Chem.2008,282(1),117.
(17) Li,Y.;Zhang,J.Laser&Photonics Rev.2010,4(4),517.
(18) Hashimoto,K.;Kawai,T.;Sakata,T.Chem.Lett.1983,12(5),709.
(19)Misawa,H.;Sakuragi,H.;Usui,Y.;Tokumaru,K.Chem.Lett.1983,7,1021.
(20) Li,Y.X.;Guo,M.M.;Peng,S.Q.;Lu,G.X.;Li,S.B.Int.J.Hydrog.Energy 2009,34(14),5629.doi:10.1016/j.ijhydene.2009.05.100
(21)Abe,R.;Hara,K.;Sayama,K.;Domen,K.;Arakawa,H.J.Photochem.Photobiol.A:Chem.2000,137(1),63.doi:10.1016/S1010-6030(00)00351-8
(22) Shimidzu,T.;Iyoda,T.;Koide,Y.J.Am.Chem.Soc.1985,107(1),35.doi:10.1021/ja00287a007
(23)Kalyanasundaram,K.;Kiwi,J.;Grätzel,M.Helv.Chim.Acta 1978,61,2720.
(24) Islam,S.D.M.;Konishi,T.;Fujitsuka,M.;Ito,O.;Nakamura,Y.;Usui,Y.Photochem.Photobiol.2000,71(6),675.doi:10.1562/0031-8655(2000)071<0675:PROMVU>2.0.CO;2(25) Keller,V.;Bernhardt,P.;Garin,F.J.Catal.2003,215(1),129.doi:10.1016/S0021-9517(03)00002-2
(26) Zhang,W.;Hong,J.;Zheng,J.;Huang,Z.Y.;Zhou,J.R.;Xu,R.J.Am.Chem.Soc.2011,133(51),20680.doi:10.1021/ja208555h
(27)Zheng,Z.K.;Huang,B.B.;Qin,X.Y.;Zhang,X.Y.;Dai,Y.;Whangbo,M.H.J.Mater.Chem.2011,21(25),9079.doi:10.1039/c1jm10983a
(28) Pelet,S.;Grätzel,M.;Moser,J.E.J.Phys.Chem.B 2003,107,3215.
(29) Dürr,H.;Boβmann,S.;Beuerlein,A.J.Photochem.Photobiol.A:Chem.1993,73,233.doi:10.1016/1010-6030(93)90010-I
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
汽车工程师(2021年12期)2022-01-18
世界科学技术-中医药现代化(2021年8期)2021-12-21
世界科学技术-中医药现代化(2021年7期)2021-11-04
古今农业(2021年2期)2021-08-14
陶瓷学报(2019年5期)2019-01-12
无机盐工业(2017年5期)2017-05-25
郑州大学学报(理学版)(2017年1期)2017-04-07
小溪流(画刊)(2017年3期)2017-03-23
化工管理(2017年25期)2017-03-05