以烟酸为配体的铜配合物的合成、晶体结构及磁学性质
2013-09-15 03:04:06刘光祥
无机化学学报 2013年9期
刘光祥
(南京晓庄学院生物化工与环境工程学院,南京 211171)
0 Introduction
The rational d esign and controlled synthesis of metal-organic frameworks (MOFs)has become an exciting and expanding approach to novel functional materials in the realm of crystal engineering and supramolecular chemistry[1].Great interest has been focused on not only fine-tuning their structural diversities[2]and interesting topologies[3]but also their potential applications as functional materials in many areas,such as photoluminescence[4],magnetism[5],catalysis[6],gasstorage[7],conductivity[8],non-linear optics(NLO)[9], ion exchange[10], ferroelectricity[11],optoelectronic effects[12],and spin-transition behaviour[13].The vast majority of work involves the use of polyfunctional organic ligands bonded to d-block transition metal atoms by metal coordination-directed self-assembly processes,which lead to the construction of hybrid frameworks with fascinating topologies.However,some major challenges in this research field,such as,(i)how to control polymer dimensionality;(ii)how to improve framework stability;(iii)how to design novel structural compounds with desirable properties;(iv)how tosynthesisdesirablecompoundsin convenient ways,still remain.
Recent elaboration has demonstrated the dramatic influence of organic components on the framework of inorganic materials,such as how organic components modify the inorganic frameworks[14].Nicotinic acid(Hnic),which has two types of coordinating atoms(N and O),can be used as an electron transfer mediator or chelate ligand[15-19].The flexibility of the coordination sphere of nicotinic acid around transition metal ions,in combination with stereo and crystal packing forces,usually leads to tremendous network diversity.Taking advantage of some interesting properties of nicotinic acid and hydrothermal reactions,we present the synthesis,structural characterization,and magnetic properties of a new coordination polymer based on nicotinic acid,[Cu2(nic)4(H2O)]n(1).
1 Experimental
1.1 Materials and general methods
All reagents for syntheses and analyses were purchased from commercial sources and used as received without further purification.Elemental analyses(C,H and N)were performed on a Vario ELIII elemental analyzer.Infrared spectra were performed on a Nicolet AVATAR-360 spectrophotometer with KBr pellets in the 400~4 000 cm-1region.The X-band EPR spectra were recorded on a Bruker 2000-D-SRC spectrometer on polycrystalline material at room temperature.Variable-temperature susceptibilities were measured using a MPMS-7 SQUID magnetometer.Diamagnetic corrections were made with Pascals constantsfor all constituent atoms[20].
1.2 Synthesisof[Cu2(nic)4(H 2O)]n(1)
A mixture of Hnic(0.4 mmol,49.2 mg),CuCl2·2H2O(0.2 mmol,34.0 mg)and H2O(15 mL)was stirred for 30 min,and the pH value of the solution was adjusted to about 5 with triethylamine.After stirring for another 30 min,the mixture was transferred to a 25 mL Teflon-lined stainless steel vessel and heated at 120℃for 2 d.Then the reaction system was cooled to room temperature,and blueblock crystalsof 1 were obtained.Yield:62% (based on Hnic).Anal.Calcd.for C24H18N4O9Cu2(%):C,45.50;H,2.86;N,8.84.Anal.Found(%):C,45.45;H,2.88;N,8.89.IRspectrum:3 651,3 062,1 611,1 584,1454,1 404,1 381,1 322,1 260,1 192,1 155,1 089,1 057,998,882,826,701,643,575 and 545 cm-1.
1.3 X-ray crystallography
The single crystals of complex 1 with approximate dimensions of 0.40 mm×0.40 mm×0.40 mm was placed on an Oxford Xcalibur Gemini Ultra diffractometer.The diffraction data were collected using a graphite monochromated Mo Kα radiation(λ=0.071 073 nm)at 290(2)K.Data reduction and empirical absorption correction were performed using the CrysAlisPro program[21].The structure was solved by the direct method using SHELXS-97[22]and refined by full-matrix least squareson F2using SHELXL-97[23].All of the nonhydrogen atoms were refined anisotropically.The hydrogen atoms were generated theoretically onto the specific atoms and refined with isotropic thermal parameters riding on the parent atoms.In addition,PLATON/SQUEZEE routine was used for their disordered solvent molecules in view of helps in crystallographic justification[24].The details of the crystal parameters,data collection and refinement for 1 are summarized in Table 1,and selected bond lengths and angles with their estimated standard deviations are listed in Table 2.
CCDC:921590.
2 Resultsand discussion
2.1 Crystal structure
Single-crystal X-ray diffraction of 1 reveals a complicated 3D coordination polymer that crystallizes in the monoclinic space group P21/c.The asymmetric unit of 1 contains two crystallographically unique Cu(Ⅱ)ions,four individual nic anions and one coordinated water molecule.As shown in Fig.1,each of the two crystallographically distinct Cu(Ⅱ)ions displays slightlydistorted octahedral[CuN2O4]coordination geometry.The arrangement of donor atoms about each of the two Cu(Ⅱ)ions is identical,albeit with subtly altered bond distances and angles.The two nitrogen donors,one belonging to an exobidentateμ2-nic anion and the other to an exotridentateμ3-nic anion,are disposed cis relative to each other.Oxygen atoms belonging to two differentμ3-nic anions are also situated in a cis orientation.The remaining two coordination sites occupied by oxygen atoms from aμ2-nic anion and aμ2-water molecule,again in a cis disposition.The μ3-nic pyridyl nitrogen donor is arranged trans to theμ2-water molecule that bridge each pair of Cu(Ⅱ)ions(Cu-O-Cu angles=114.44(12)°)into binuclear [Cu(μ-H2O)(μ3-
nic)2Cu]2+units with a Cu-Cu distance of 0.355 5 nm.The Cu-Odistancesrange from0.202 0(2)to 0.212 3(3)nm,and the Cu-N distances are between 0.208 1(3)and 0.211 4(3)nm.The O(N)-Cu-O(N)bond angles range from 83.69(10)°to 177.49(11)°,which are similar to thoseobserved in thereported complexes[25].

Table 1 Crystal data and structure refinement for 1

Table 2 Selected bond lengths(nm)and angles(°)for 1

Scheme 1 Coordination modes of nic anion in 1

Fig.1 Coordination environment of Cu(Ⅱ)in complex 1 with thermal ellipsoids at 30%probability

Fig.2 Views of helical 1-D chain ofμ3-nic connected binuclear units in 1 along the c axis
As exhibited in Fig.2,each binuclear unit is turn connected to its neighbors throughμ3-nic anions to form 1D [Cu2(μ-H2O)(μ3-nic)2]helical chains that run down the c axis.The structure of 1 also contains rippled 2D cationic[Cu4(μ-H2O)2(μ2-nic)4]layerscoincident with the bc plane,built fromthe junction of the two typesof[Cu2(μ-H2O)]4+subunits through all of the μ2-nic anions(Fig.3).This 2D layer stands in contrast to the [Co2(μ-H2O)(μ2-nic)2]1D double chain staircase motif observed in the 3D open framework coordination polymer [Co2(-H2O)(μ2-nic)4],which contains similar binuclear units[26].The 2D cationic layers in 1 are then stitched into the full 3D crystal structure through the aforementioned helical 1D[Cu2(μ-H2O)(μ3-nic)2]chains.
Better insight into such elegant frameworks can be achieved using the topology method:Firstly,based on the considerations of their connectivity,the binuclear Cu2is viewed to be five-connected nodes(Fig.4);secondly,the CuO/N coordination bonds and nic ligands are simplified to be linear connectors;then the combination of nodes and connectors suggests the 5-connected frameworks for the present case.The topological notation for such 5-connected frameworks is(44·66)net(Fig.5).In the literature[27-29],the uninodal 4-,6-and 8-connected frameworks are often encountered,but we are not aware of a precedent characterized by such topology like that found in 1.

Fig.3 View of the undulating 2D network in 1 comprised ofμ2-nic connected binuclear units

Fig.4 View of the linkages of a binuclear unit with four adjacent binuclear units in 1

Fig.5 Topological illustration for the uninodal 5-connected(44·66)net of 1
2.2 IR spectra
The absence of such bands at around 1700 cm-1in the IRspectrumindicatesthecomplete deprotonation of nicotinic acid.The characteristic bands of carboxyl groups are shown in the range of 1 584~1 611 cm-1for antisymmetric stretching and 1381 cm-1for symmetric stretching.The separations(Δν)between νasym(CO2)and νsym(CO2)bands indicate the presence of different coordination modes.The bands in the region 643~1 322 cm-1are attributed to the-CH-in-plane or out-of-plane bend,ring breathing,and ring deformation absorptions of pyridyl ring,respectively.Weak absorptions observed at 3 062 cm-1can be attributed to νC-Hof benzene ring.The IR spectra exhibit the characteristic peaks of water molecule at 3 651 cm-1[30].
2.3 Magnetic propertiesand EPR
The χMT product has a value of 0.77 cm3·K·mol-1at 300 K,well in agreement with the expected value for two non-interacting Cu(Ⅱ) ions with S=1/2 and g=2.0(Fig.6).As the temperature decreases,so does the χMT product,indicating antiferromagnetic coupling between the Cu(Ⅱ) ions in 1.Accordingly,a maximum is seen in the susceptibility at 35 K,indicating the transition to a diamagnetic state below this temperature.As the temperature decreases further a plateau at aχMT value of 0.045 cm3·K·mol-1is observed,as well as a rise in the susceptibility,clear indication of a small amount of paramagnetic impurity,probably a Cu(Ⅱ)monomer or salt.

Fig.6 Temperature dependence ofχMT(○)and χM(□)for 1
According to the crystal structure,complex 1 can be considered as binuclear from the viewpoint of magnetism,in which two Cu(Ⅱ)ions are linked by two carboxylate and oneμ2-water moleculebridgessince the coupling through nic anions can almost be negligible.Taking into account the binuclear Cu(Ⅱ)model,the magnetic susceptibility data are analyzed by using the familiar Bleaney-Bowers expression based on a Heisenberg Hamiltonian H=JS1·S2[20]:
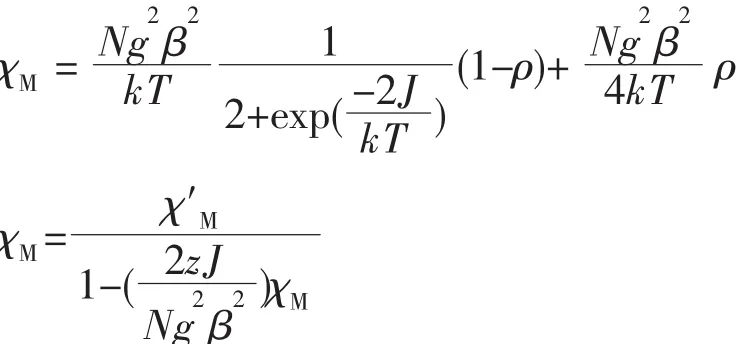
where N,g,b and k have their usual meanings.ρis a variable fraction of paramagnetic impurities.zJ accounts for the interdimer interactions.The leastsquares analysis of magnetic susceptibility data led to J=-2.63 cm-1,zJ=0.041 cm-1,g=2.10,ρ=8.1%,and R=7.23×10-5.The fitting results indicate that the magnitude of the magnetic coupling between the adjacent Cu(Ⅱ)ions in 1 is very weak.As is well known,the magnetic properties are closely related to the bridging conformation adopted by the carboxylate group in these polynuclear systems,the syn-syn carboxylato bridge is able to mediate very strong antiferromagnetic interactions in Cu(Ⅱ)complexes,whereas the syn-anti type will normally result in a weak magnetic exchange interaction[31].In 1,the carboxylate groups of nic anion adopt a syn-anti fashion to link the Cu(Ⅱ)ions and the resulting magnetic exchanges isweak.
The powdered EPR spectra of 1 were measured at roomtemperature and the spectra are shown in Fig.7.It is all typical Cu(Ⅱ)signals having the character of axial eld.The g∥and g⊥values are 2.225 and 2.078,respectively.The order of g∥>g⊥>2.0023 matches to all the Cu(Ⅱ)complexesthat the unpaired electron exists intetragonal or elongated octahedral Cu(Ⅱ)coordination sphere.While the broadened signal of 1 with the G value of 3.48 (less than 4) indicates an antiferromagnetic interaction between the two Cu(Ⅱ)ions of the binuclear complex[33].Similar magnetic and EPRdatawereobserved for other copper(Ⅱ)dimer[34].
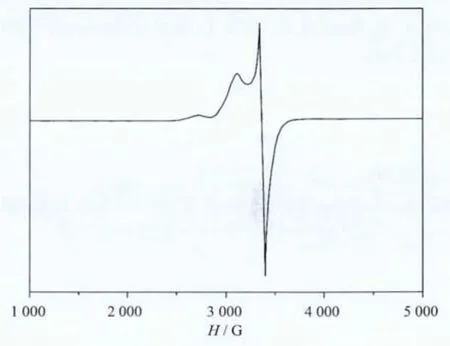
Fig.7 EPR spectra of 1 at room temperature
[1]Robin A Y,Fromm K M.Coord.Chem.Rev.,2006,250:2127-2157
[2]Hou JZ,Li M,Li Z,et al.Angew.Chem.Int.Ed.,2008,47:1711-1714
[3]Blatov V A,Carlucci L,Ciani G,et al.CrystEngComm,2004,6:378-395
[4]Zou R Q,Zhong R Q,Du M,et al.Cryst.Growth Des.,2008,8:452-459
[5]Li J R,Yu Q,Saöudo E C,et al.Chem.Mater.,2008,20:1218-1220
[6]Gomez-Lor B,Gutiörrez-Puebla E,Iglesias M,et al.Inorg.Chem.,2002,41:2429-2432
[7]Luo J,Xu H,Liu Y,et al.J.Am.Chem.Soc.,2008,130:9626-9627
[8]Coronado E,Day P.Chem.Rev.,2004,104:5419-5448
[9]Cariati E,Macchi R,Roberto D,et al.J.Am.Chem.Soc.,2007,129:9410-9420
[10]Mi L,Hou H,Song Z,et al.Chem.-Eur.J.,2008,14:1814-1821
[11]Bujak M,Angel R J.J.Solid State Chem.,2005,178:2237-2246
[12]Chen W,Yuan H M,Wang J Y,et al.J.Am.Chem.Soc.,2003,125:9266-9267
[13]Kahn O,Martinez CJ.Science,1998,279:44-48
[14]Hagrman P J,Hagrman D,Zubieta J.Angew.Chem.Int.Ed.,1999,38:2639-2684
[15]Liu F C,Zhao J P,Hu B W,et al.Dalton Trans.,2010,39:1185-1187
[16]Cheng JW,Zheng ST,Yang G Y.Inorg.Chem.,2008,47:4930-4935
[17]Zhang J,Kang Y,Zhang J,et al.Eur.J.Inorg.Chem.,2006:2253-2258
[18]Yeh C W,Suen M C,Hu H L,et al.Polyhedron,2004,23:1947-1952
[19]Liu F C,Zeng Y F,Zhao J P,et al.Inorg.Chem.,2007,46:7698-7700
[20]Kahn O.Molecular Magnetism.New York:VCH Publishers,Inc.,1993.
[21]CrysAlisPro,Oxford Diffraction Ltd.,Oxfordshire,U.K.,2010.
[22]Sheldrick G M.SHELXS-97,Program for Crystal Structure Solution,University of Göttingen,Germany,1997.
[23]Sheldrick G M.SHELXL-97,Program for the Refinement of Crystal Structure,University of Gttingen,Germany,1997.
[24]Köppers H,Lieba F,Spek A L.J.Appl.Crystallogr.,2006,39:338-346
[25]Gurunatha K L,Maji T K.Eur.J.Inorg.Chem.,2009:1592-1599
[26]Liu Y H,Tsai H L,Lu Y L,et al.Inorg.Chem.,2001,40:6426-6431
[27]Jiang X R,Yuan H Y,Feng Y L.J.Solid State Chem.,2012,191:181-190
[28]Chen S S,Zhao Y.Fan J,et al.CrystEngComm,2012,14:3564-3576
[29]Gong Y,Li J,Qin J B,et al.Cryst.Growth Des.,2011,11:1662-1674
[30]Nakamoto K.Infrared and Raman Spectra of Inorganic and Coordinated Compounds.5th Ed.New York:Wiley&Sons,1997.
[31]Colacio E,Ghazi M,Kivekas R,et al.Inorg.Chem.,2000,39:2882-2890
[32]Kasumov V T,Köksal F,Sezer A.Polyhedron,2005,24:1203-1211
[33]Psomas G,Raptopoulou C P,Iordanidis L,et al.Inorg.Chem.,2000,39,3042-3048
[34]Segla P,Palicova M,Miklos D,et al.Z.Anorg.Allg.Chem.,2004,630:470-478
猜你喜欢
科技进步与对策(2023年16期)2023-09-01 07:09:24
科技进步与对策(2022年23期)2022-12-27 12:57:12
土壤学报(2022年4期)2022-10-22 05:39:08
蜜蜂杂志(2022年5期)2022-07-20 09:54:12
蜜蜂杂志(2022年2期)2022-04-15 03:08:40
科技进步与对策(2021年20期)2021-10-30 03:02:02
蜜蜂杂志(2021年3期)2021-10-19 10:01:28
科技进步与对策(2020年10期)2020-06-30 02:16:22
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21 05:32:46
塑料助剂(2019年3期)2019-07-24 08:51:24