
Th17细胞分化的转录调控网络①
2013-07-30 13:33哈尔滨医科大学附属第二医院神经内科三病房哈尔滨150086
中国免疫学杂志 2013年5期
徐 丹 杨 丹 付 锦 (哈尔滨医科大学附属第二医院神经内科三病房,哈尔滨150086)
初始T细胞通过其表面的T细胞抗原受体识别病原微生物,受不同共刺激分子的调节,活化各种信号转导通路,最终分化为多种效应性T细胞,发挥抗炎、调节免疫等作用。经典的根据初始T细胞所分泌细胞因子种类和功能的不同,将其分为辅助性T细胞1型(T help cell 1,Th1)、2型(T help cell 2,Th2)和调节性 T(Treg)细胞,Th1和 Th2细胞分别参与细胞和体液介导的免疫应答,Treg细胞可以抑制免疫反应。近来,研究报道一种新的T细胞系,因其能特异性的分泌产生IL-17细胞而被称为Th17细胞,Th17细胞通过孤核受体 γt(Retinoic acid receptor-related orphan receptorγ t,RORγt)介导的IL-17的产生调节炎症性自身免疫性疾病[1]。
1 Th17细胞分化的调节网络
白介素6(Interleukin-6,IL-6)与转化生长因子(Transforming growth factorβ,TGFβ)可通过 JAKSTAT3通路激活转录因子RORγt促进IL-17的表达,在这一过程中,转录因子孤核受体 RORγt(RORα)、信号转导子和转录激活子3(Signal transducer and activator of transcription 3,STAT3)、活化 T细胞核因子(Nuclear factor of activated T cells,NFAT)、B细胞激活转录因子 (B-cell activating transcription factor,BATF)与IL-17基因启动子直接结合调节Th17细胞分化;而干扰素调节因子4(Interferon regulatory factor4,IRF4)、STAT3、BATF 与RORγt基因Rorc结合间接调节Th17细胞分化;还有些转录因子如:Th1相关转录因子 (Th1-associated transcription factor,T-bet),表皮脂肪酸结合蛋白(Epidermal-Fatty acid-binding protein,E-FABP),芳香烃受体(Aryl hydrocarbon receptor,AHR),Ets转录因子家族成员Ets-1(E26 tansformation-specific 1),插头框蛋白p3(Forkhead box P3,FoxP3)和过氧化物酶体增生物激活受体γ(Peroxisome proliferator-activated receptorγ,PPARγ),虽然不能与 IL-17 基因或Rorc直接结合,但对Th17细胞分化也具有重要的间接调节作用。下面我们将具体阐述这些转录因子对Th17细胞分化的调节作用。
1.1 Th17细胞分化的直接调控
1.1.1 孤核受体 γt(RORγt) 每个T细胞亚群都有相对特异性的Marker,是其分化的重要转录调控因子,如Th1细胞为T-bet,Th2细胞为 GATA3,Treg细胞为Foxp3,孤核受体γt被认为是调节Th17细胞分化的重要转录因子。体外实验中,用 IL-6和TGFβ诱导IL-17时需要RORγt,而且即便在没有外源性细胞因子诱导的情况下,RORγt的充分表达足以诱导 IL-17的表达。IL-17启动子上有许多RORγt的结合位点,通过染色体免疫沉淀法及测序(Chromatin immunoprecipitation followed by massive parallel sequencing,ChIP-Seq)证实 RORγt能结合IL-17的基因。RORγt由Rorc基因启动子起始位点的基因突变导致其编码的氨基酸序列改变而产生。另一维甲酸受体家族成员RORα也可促进Th17的分化,RORα由Rora基因编码,但敲除Rora基因仅轻度降低IL-17的产生,只有当同时敲除Rora和Rorc基因,才能完全抑制IL-17的产生[2]。
1.1.2 信号转导子和转录激活子3(STAT3) 已知细胞因子IL-6、IL-21和IL-23能激活不同的受体复合体,使下游基因Jak1、Jak2和Tyr2磷酸化,活化细胞内的STAT3,在STAT3基因敲除的T细胞中,IL-21、IL-23受体和 IL-17的表达明显降低,表明STAT3对Th17细胞分化具有重要调节作用。通过ChIP-Seq不仅证实STAT3可结合IL-17A、IL-21和IL-17F基因启动子及IL-17A和IL-17F位点的基因间区域,还发现STAT3能结合Rorc基因[3]。敲除STAT3使RORγt的产生受损,而此时即便使RORγt过表达也仅能部分恢复 IL-17的表达,这表明RORγt可能在STAT3的下游发挥作用。RORγt和STAT3可单独或形成复合体共同结合于IL-17基因位点,从而最大限度地诱导IL-17的产生。
1.1.3 活化T细胞核因子(BATF)BATF为核碱性亮氨酸拉链蛋白,它属于AP-1/ATF转录因子超家族。活化的Th细胞亚群(包括Th1、Th2和Th17细胞)中,BATF基因转录明显增加。在体内外实验中,BATF缺乏将导致初始CD4+T细胞不能产生IL-17,但可产生Treg细胞,同时 RORγt和 RORα的表达也减少,从而保护小鼠不发生实验性自身免疫性脑炎(EAE)。研究已证实,BATF能直接结合到Rora和 Rorc基因启动子域或基因间区域,维持RORγT和RORα的表达,从而促进Th17细胞相关的细胞因子的表达[4]。另外,BATF能结合IL-17基因启动子诱导IL-17的表达。
1.1.4 活化T细胞核因子(NFAT)NFAT是位于迁移细胞胞质中的一组钙依赖的转录因子,对T细胞免疫调节具有重要作用。NFAT不同亚型对T细胞的调节作用不同。初始T细胞的活化需要共刺激分子,后者可激活NFAT、AP-1或NF-κB信号通路。已知IL-6可促进Th17细胞分化,IL-6受体活化可诱导NFAT1的转录,增加了NFAT1对T细胞受体刺激反应的活性;IL-6还可诱导IL-21的表达,IL-21基因启动子有许多NFAT的结合位点。在促进小鼠T细胞向Th17细胞分化的条件下,NFAT1的活化可促进IL-17和IL-10的产生,这与NFAT1可直接结合IL-17和IL-10基因位点的远端调节域有关。尽管IL-17产生增加,但并未增加EAE的易感性,这与免疫调节因子IL-10有利于Treg细胞的分化,从而抑制炎症反应有关[5]。人类IL-17A基因近端启动子包含两个NFAT结合位点,它们对IL-17的调节可能有重要作用。
1.2 Th17细胞分化的间接调控
1.2.1 干扰素调节因子4(IRF4)IRF4最初被认为是促进Th2细胞分化的转录因子,近来发现IRF4可能与 STAT3共同诱导RORγt的表达,在 RORγt的上游发挥转录调节作用[6],缺乏IRF4的T细胞仅表达少量的 RORγt,即便上调 RORγt表达,也只能部分恢复Th17细胞的分化。此外,IRF-4与干扰素调节因子4结合蛋白(IBP)相互作用可阻止IRF4与IL-17和IL-22基因的靶向结合,IBP缺乏时IL-17和IL-22的表达增加,从而促进Th17细胞的分化。还有研究认为,IL-1可激活IRF4并促进Th17细胞的早期分化,但其调节机制还有待研究证实。
1.2.2 Th1相关转录因子 (T-bet) 有报道认为T-bet对T细胞分化的作用受T细胞信号强度的影响。强的TCR信号上调T-bet,而弱的TCR信号有利于IL-4和IL-17的产生。Mathur等研究显示T-bet/记忆性初始CD4+T细胞更倾向于对IL-23作出反应产生IL-17,T-bet正向调节IL-23受体的表达,而IL-23可促进Th17分化。但Lazarevic等研究发现T-bet与Runx1相互作用,阻止Runx1(Runt相关转录因子)活化Rorc基因,而Runx1可通过活化Rorc基因上调 IL-17 的表达[7,8],因此,认为 T-bet可能通过Runx1抑制Th17细胞分化。
1.2.3 Ets转录因子家族成员 ETS-1 ETS-1是ETS转录抑制家族成员之一,在造血、血管发生和肿瘤的发展中发挥重要作用[9]。最近研究表明ETS-1与自身免疫性疾病的发生相关[10]:当ETS-1缺失会导致Th17细胞分化增加,然而ETS-1未与IL-17基因启动子直接结合,此抑制作用为间接作用。有研究显示这种间接的负向调节作用与IL-2相关,IL-2是一种体外的T细胞生长因子,ETS-1缺失后降低IL-2产生,IL-2能通过活化STAT5促进Treg细胞的分化而抑制Th17细胞的分化。
1.2.4 芳香烃受体(AHR)AHR是配体依赖的转录因子,AHR与芳香受体核转运蛋白(ARNT)结合成二聚体调节基因的表达[11]。FICZ是一种色氨酸源性的光合产物,是对AHR有高亲和力的内源性配体,两者结合激活 AHR,促进 IL-22、IL-17A和 IL-17F的表达。有研究显示AHR可通过干扰STAT1的活化来调节Th17细胞分化。AHR缺乏与STAT1的持续磷酸化有关,而STAT1可作为抑制子结合IL-17基因启动子,且STAT1还能干扰ROR蛋白与IL-17基因启动子的相互作用,因此设想AHR可能是通过抑制细胞因子IFN-γ和IL-27活化STAT1从而促进Th17细胞分化的[12]。另有一种人工合成配体2,3,7,8-四氯二苯并-对-二恶英(TCCD),与 AHR结合后可以促使活化的CD4+T细胞分化为Treg细胞,抑制免疫反应。目前尚不清楚AHR是如何调节Th17细胞的分化,但却使我们认识到环境毒素对自身免疫性疾病发病机制的影响。
1.2.5 过氧化物酶体增生物激活受体γ(PPARγ)
PPARγ是脂质活化的转录因子,与相应配体结合后通过诱导靶基因脂肪酸结合蛋白(FABP)的表达来调节脂肪细胞的分化和脂质代谢。近来发现PPARγ可调节Th细胞的克隆,并因其可负向干扰抗炎细胞信号转导而具有抗炎作用。研究显示PPARγ是Th17细胞分化的固有抑制子,PPARγ的活化可以阻止抑制子复合体从RORγT基因启动子的去除,抑制RORγt的表达及RORγt诱导的 Th17细胞的分化[13]。药理活性的PPARγ抑制维甲酸和甲状腺素受体辅阻遏物从RORγt启动子的去除,从而干扰RORγt的转录。
1.2.6 脂肪酸结合蛋白(FABP)FABP是一种脂类伴侣蛋白。研究发现表皮的FABP(E-FABP)和脂肪的FABP(A-FABP)均能调节巨噬细胞和树突细胞的炎症反应。其中,A-FABP表达于巨噬细胞、树突细胞和脂肪细胞中[14],而 E-FABP在各种FABP家族成员中均有表达,并且可以表达于CD4+T细胞中。研究显示E-FABP可通过Foxp3/RORγT(RORα)来调节Th17细胞分化[15]。与野生型鼠比较,E-FABP缺乏鼠的CD4+T细胞中Foxp3的表达增加,而RORγT和RORα表达减少。另外,E-FABP缺乏的CD4+T细胞中PPARγ表达增加致IL-17的产量减少,这一过程可被PPARγ的拮抗剂GW9662逆转。更多的E-FABP的分子机制还有待进一步实验研究。
2 细胞因子在Th17细胞分化中的作用
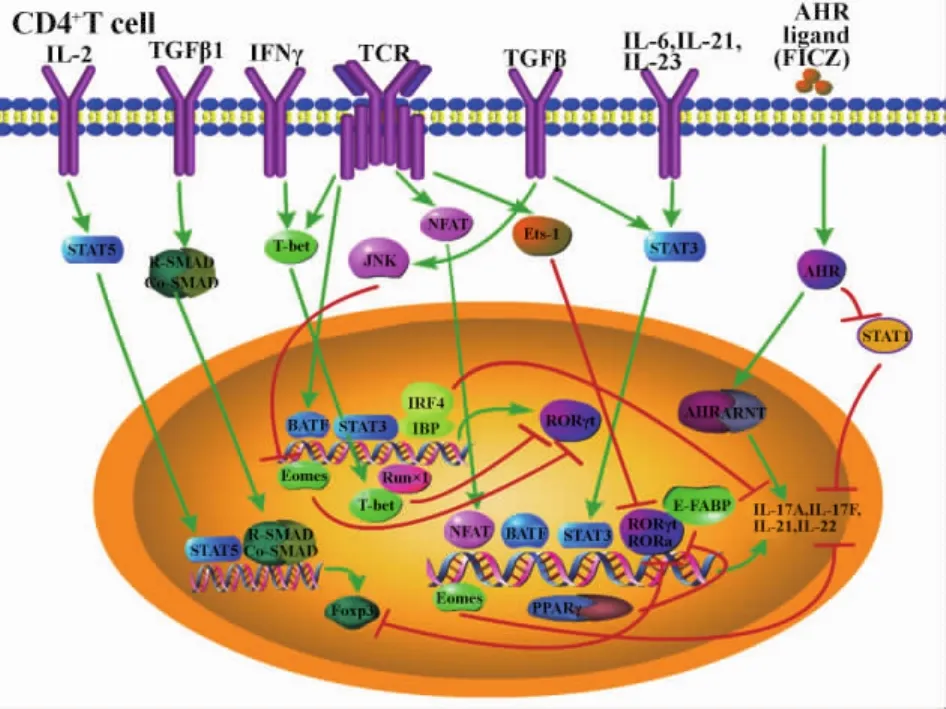
图1 .Th17细胞分化的信号转导通路Fig.1 Signal transduction pathway in the control of Th17 cell
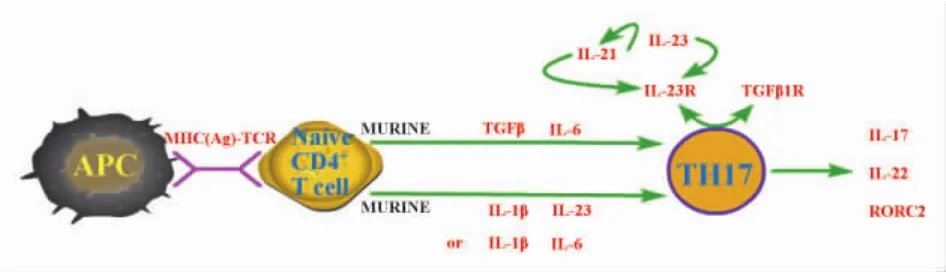
图2 .细胞因子对Th17细胞分化的调节Fig.2 Cytokine regulation of Th17 cells differentiation
2.1 IL-6和 TGFβ IL-6是促进初始 T细胞向Th17分化的重要细胞因子[16],IL-6联合TGFβ可有效地诱导初始T细胞产生IL-17。然而IL-6缺乏时仅降低了Th17细胞的数量,表明还有其他细胞因子可以替代IL-6,由多种细胞分泌产生的细胞因子IL-21可联合TGFβ在缺乏IL-6时诱导IL-17的表达。而且,IL-6可诱导初始T细胞分泌IL-21,IL-21还能通过自分泌的方式诱导自我表达、促进IL-23受体的产生及抑制IFNγ的产生,从而维持初始T细胞向Th17细胞分化。但在活体实验中Th17细胞分化不受IL-21信号的影响。
TGFβ以浓度依赖的方式调节Th17促炎/Treg抗炎的两极分化[17]。具体来说,在有促炎因子IL-6和IL-21存在的前提下,低浓度的TGFβ促进IL-23受体的表达,同时,IL-6以STAT3依赖的方式抑制Foxp3的表达,从而促进Th17细胞分化。相反,高浓度的TGF-β降低IL-23受体的表达,增加Foxp3的表达而促进Treg细胞分化。Smad蛋白(drosophila mothers against decapentaplegic protein)通路是TGFβ一个重要信号转导通路,敲除冠蛋白1基因可通过损害TGFβ受体介导的Smad3信号通路促进CD4+T细胞向Th17细胞分化。TGFβ/Smad2信号通路可促进类浆细胞树突细胞向初始T细胞极化,并向Th17细胞分化。但最近有研究显示TGFβ调节Th17分化不依赖于Smad通路,而是通过JNK信号通路抑制转录因子 Eomes的表达,Eomes能与Rorc和IL-17A基因启动子近端结合发挥抑制作用,从而促进Th17细胞分化[18]。另有研究显示活化的Th17细胞可通过自分泌TGFβ1的方式促进Th17细胞表型的稳定[19]。但是人Th17细胞的分化不需要TGFβ,取而代之的是IL-1β联合IL-6或IL-23,它们能够上调孤核受体基因 Rorc2,促进 IL-17的分泌。
2.2 IL-23 长期以来IL-23被认为是IL-17产生的重要诱导子,然而近来研究发现初始T细胞并不表达IL-23受体,且它需要被诱导产生,故现在认为IL-23不能单独诱导IL-17的分泌。Th17细胞可表达IL-23受体,IL-21和IL-23都可诱导IL-23受体的表达,一旦IL-23受体的表达上调,IL-23即可联合TGFβ诱导IL-17的表达。另外,在自身免疫性关节炎的小鼠研究中发现IL-23可通过降低T-bet和Foxp3表达,增加IL-17A、IL-17F和IL-22的水平,从而促进Th17细胞分化。这些表明Th17细胞分化的最初阶段不需要IL-23,但是IL-23在之后的快速上调对Th17细胞的扩增具有重要作用,即 IL-23在Th17细胞分化后期发挥重要作用。近来,研究显示IL-23可诱导粒细胞巨噬细胞集落刺激因子的产生促进 Th17 细胞分化[20]。
3 结束语
综上所述,不同的T细胞受体、细胞因子及转录因子对Th17细胞分化的调节并不是简单的线性关系,而是形成复杂的调控网络,共同影响Th17细胞的分化、成熟及功能的发挥,但完整的调节机制及其之间的相互关联和影响仍需进一步研究证实。
1 Chen SJ,Wang Y L,Fan H C et al.Current status of the immunomodulation and immunomediated therapeutic strategies for multiple sclerosis[J]. Clinical and Developmental Immunology, 2012;2012:970789.
2 Yang X O,Pappu B P,Nurieva P et al.Thelper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma[J].Immunity,2008;28:29-39.
3 Durant L,Watford W T,Ramos H L et al.Targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis[J].Immunity,2010;32:605-615.
4 Schraml B U,Hildner K,Lse W et al.The AP-1 transcription factor Batf controls Th17 differentiation[J].Nature,2009;460:405-409.
5 Ghosha S,Koralova SB,Stevanovica I et al.Hyperactivation of nuclear factor of activated T cells 1(NFAT1)in T cells attenuates severity of murine autoimmune encephalomyelitis[J].Immunology,2010;107(34):15169-15174.
6 Mudter J,Yu J L,Zufferey C et al.IRF4 regulates IL-17A promoter activity and controls RORct dependent Th17 colitis in vivo[J].Inflamm Bowel Dis,2011;17(6):1343-1358.
7 Oestreich K J,Weinmann A S.T-bet employs diverse regulatory mechanisms to repress transcription[J].Trends in Immunology,2012;(2):78-83.
8 Lazarevic V,Chen X,Shim JH et al.T-bet represses Th17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat[J].Nat Immunol,2011;12:96-104.
9 Arderiu G,Pena E,Aledo R et al.Ets-1 transcription is required in tissue factor driven microvessel formation and stabilization[J].Angiogenesis,2012;15(4):657-669.
10 Pan H F,Leng R X,Tao JH et al.Ets-1:a new player in the pathogenesis of systemic lupus erythematosus?[J].Lupus,2011;20:227-230.
11 Lo R,Matthews J.High-resolution genome-wide mapping of AHR and ARNT binding sites by ChIP-Seq[J].Toxicol Sci,2012;130(2):349-361.
12 Kimura A,Naka T,Nohara K et al.Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells[J].Immunology,2008;105:9721-9726.
13 Klotz L,Burgdorf S,Dani I et al.The nuclear receptor PPARγ selectively inhibits Th17 differentiation in a Tcell-intrinsic fashion and suppresses CNS autoimmunity[J].Brief Definitive Repert,2009;206(10):2079-2089.
14 He J,Tian Y,Li J J et al.Expression pattern of adipocyte fatty acid-binding protein gene in different tissues and its regulation of genes related to adipocyte differentiation in duck[J].Poult Sci,2012;91(9):2270-2274.
15 Li B,Reynolds JM,Stout R D et al.Regulation of Th17 differentiation by epidermal fatty acid-binding protein[J].Immunol,2009;182:7625-7633.
16 Camporeale A,Poli V.IL-6,IL-17 and STAT3:a holy trinity in auto-immunity? [J]. Frontiers in Bioscience, 2012;17:2306-2326.
17 Hatton R D.TGF-β in Th17 cell development:The truth is out there[J].Immunity,2011;34:288-290.
18 Ichiyama K,Sekiya T,Inoue N et al.Transcription factor smad-independent T helper 17 cell induction by transforming-growth factor-β is mediated suppression of eomesodermin[J].Immunity,2011;34:741-754.
19 Gutcher I,Donkor M K,Ma Q et al.Autocrine transforming growth factor-β1 promotes in vivo Th17 cell differentiation[J].Immunity,2011;34:396-408.
20 Codarri L,Gyülvészi G,Tosevski V et al.RORγt drives production of the cytokine GM-CSF in helper T cells,which is essential for the effector phase of autoimmune neuroinflammation[J].Nat Immunol,2011;12(6):560-567.
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
清华金融评论(2022年4期)2022-04-13
现代临床医学(2021年4期)2021-07-31
科学与财富(2021年33期)2021-05-10
国际放射医学核医学杂志(2021年10期)2021-02-28
房地产导刊(2020年7期)2020-08-24
作文成功之路·小学版(2020年6期)2020-07-27
作文成功之路·小学版(2020年5期)2020-06-11
中国外汇(2019年14期)2019-10-14
中西医结合心脑血管病杂志(2016年20期)2016-03-01