
无患子总皂苷质控品的制备
2013-05-18 07:29徐德平汪何雅姚卫蓉
食品工业科技 2013年8期
伍 恒,张 翠,翁 震,徐德平,汪何雅,姚卫蓉
(1.江南大学食品学院,江苏无锡214000;2.福建源华林业生物科技有限公司,福建三明354500)
无患子又名肥皂树,其果皮中含有丰富的无患子皂苷,是一种天然的非离子型表面活性剂,具有很强的降低表面张力的作用,泡沫丰富、手感细腻、去污力强。用从无患子果提取的皂苷制得的皂乳洗碗剂、蔬果清洗露、食品机械清洗剂等用于食品工业中不会带来萤光剂、表面活性剂、人工激素、江河湖泊富营养化等负面问题,在一定程度上保证了食品安全;另外,无患子皂苷还能迅速分解,不会对环境造成任何污染。目前,国内对无患子皂苷的研究仍处于初步阶段,市面上没有无患子皂苷的纯品出售,关于其定性研究比较困难。关于皂苷的测定方法较多,主要有紫外分光光度法、薄层色谱法、高效液相色谱法、高效液相-质谱联用法、气相色谱法、高效毛细管电泳法等,以上分析方法各个侧重点不同:比色法、紫外分光光度法比较适用于检测总皂苷含量;薄层色谱法在中药成分研究中应用较为广泛;高效液相色谱法对于皂苷的定量检测较方便,但需要昂贵的标准品做对照[1-2];液质联用法能高效准确地对皂苷定性和定量[3-8],但设备仪器价格昂贵,操作技术要求高。分光光度法已经应用于人参总皂苷、酸枣仁皂苷、黄芪总皂苷等的测定中,证明了该方法是检测总皂苷含量的方便有效地方法之一。
所以本文采用醇提-大孔树脂吸附法自制无患子总皂苷标准品,经Libermann-Buehard反应、溶血反应定性,并采用HPLC/MS方法初步鉴定无患子总皂苷成分,经香草醛-冰醋酸比色法定量无患子总皂苷标准品纯度。目前市面上涌现出越来越多的无患子皂苷产品,例如无患子蔬果清洁露、无患子洗碗露等,所得高纯度皂苷可作为工艺研究的质量控制标准品,同时也可作为食品工业上无患子皂苷产品中皂苷含量检测的标准品。填补了市面上没有高纯度无患子总皂苷出售的空白,为进一步实验研究打好基础。
1 材料与方法
1.1 材料与仪器
无患子果皮 福建源华生物科技有限公司;95%乙醇、石油醚、正丁醇、香草醛、高氯酸(70%~72%)、冰醋酸、浓硫酸 分析纯,国药集团化学试剂有限公司;AB-8大孔树脂 天津南开大学化学工厂;MCIgelCHP20P(75~150μm)柱填料 日本三菱化成公司;GF254薄层硅胶板 山东烟台芝罘化工厂;齐墩果酸标准品 中国药品生物制品检定所;日本大耳白兔 北京百尔康纳特实验兔繁育生物技术开发有限公司。
可调试移液器 Thermo Labsystems公司;PL2002型电子天平 梅特勒-托利多仪器(上海)有限公司;GEN-14型电动微型粉碎机 四川自贡渐飞机械厂;W501型恒温水浴锅 上海申顺生物科技有限公司;CQ-88-205型萃取罐 常州市特威电气自动化系统有限公司;TU-1900型双束紫外可见分光光度计 日本岛津公司;Waters Synapt G2型超高效液相色谱-质谱联用仪 美国Waters公司;R-501型旋转蒸发器上海申顺生物科技有限公司;SHBⅢ型循环水式多用真空泵 郑州长城科工贸有限公司。
1.2 实验方法
1.2.1 无患子总皂苷质控品的制备
1.2.1.1 无患子粗皂苷的提取 将无患子果皮磨碎成20目左右,称取5kg无患子粉末于提取罐中,加15kg 70%乙醇75℃下提取3h。将第一次提取液经8层纱布过滤从罐底放出。滤渣留在提取罐中进行第二、三次提取,酒精浓度、酒精用量、提取时间和温度与第一次提取相同,合并三次滤液并离心。采用旋转蒸发仪蒸发回收乙醇至浸膏状。
1.2.1.2 无患子总皂苷的分离 将上述浸膏状无患子粗皂苷分散至石油醚中萃取3次,弃去石油醚层,水相部分用正丁醇萃取5次,至正丁醇相几乎没有颜色,合并正丁醇相,减压浓缩,加入适量蒸馏水,水溶液中加入5%Ca(OH)2混匀,放置2h使沉淀,去除粘液质、鞣质和部分黄酮,抽滤后得到无患子粗皂苷溶液继续上AB-8大孔树脂柱,流速约15mL/min,上样量应低于柱体积的1/4。大孔树脂预处理、装柱及再生方法见文献[9]。先用蒸馏水洗脱去除糖类、氨基酸、色素等水溶性杂质,再用70%乙醇洗脱得无患子粗皂苷液,取出一部分干燥成粉末,HPLC-MS分析其成分。
1.2.1.3 无患子总皂苷的纯化 将大孔树脂分离所得无患子粗皂苷液浓缩,再加水稀释至浓度约5%的水溶液,上样MCI(聚苯乙烯基的反相树脂填料)凝胶柱,分别用蒸馏水、30%、70%乙醇洗脱,减压浓缩干燥得混合物Ⅰ、Ⅱ、Ⅲ,HPLC-MS、泡沫实验、TLC检测、Libermann-Buehard反应、溶血性实验同步进行定性分析。
1.2.2 无患子皂苷的定性测定
1.2.2.1 HPLC-DAD/MS检测条件 配制1mg/mL无患子粗皂苷液(大孔树脂70%洗脱部分),进行HPLCDAD/MS检测。检测条件[10-11]如下:色谱柱为BEC C18柱(2.1mm×100mm,1.7μm),流动相A:甲醇,流动相B:水,起始时40%A,60%B;20min时80%A,20%B;25min时100%A,流速0.3mL/min,柱温45℃,进样量5μL。ESI离子源;正离子模式与负离子模式相互验证,MRM多反应监测,雾化N2压力为275.8kPa,干燥N2流速9L/min,温度为350℃,毛细管电压为3.0kV,质量范围100~3000m/z,脱溶剂气温度250℃,离子源温度120℃,DAD范围190~400nm进行扫描。无患子皂苷正负离子流色谱图见图2、图3。
1.2.2.2 Libermann-Buehard反应 组分Ⅲ粉末1mg,加乙酸酐溶解,随后再加入乙酸酐-浓硫酸(1∶20)数滴,振摇[12]。观察颜色变化,颜色的变化为黄-红-紫-蓝,显示可能有三萜皂甙。
1.2.2.3 溶血反应 溶血反应是皂苷的特征反应,皂苷能导致血细胞破裂,因此可以利用溶血性来判断样品中是否含有皂苷。取大耳兔新鲜血液20mL,放入加有玻璃珠的三角瓶中,振摇10min除纤维蛋白后转至刻度离心管中,加生理盐水10mL混匀后1500r/min离心5min,除去上清液,再加适量生理盐水离心,重复至上清液无色澄清。弃去上清液取2mL红细胞,加入98mL生理盐水,即配成2%的红细胞悬浮液,备用[13]。配制混合物Ⅰ、Ⅱ、Ⅲ水溶液1mg/mL,取试管4支,分别加入滤液0.25、0.5、0.75、1mL,然后依次分别加入生理盐水2.25、2.0、1.75、1.5mL,使每一个试管中的溶液均为2.5mL,再将各试管加入2%的血细胞悬液2.5mL,振摇均匀后,立即置于37℃水浴中温浴3h,以1500r/min离心5min,若实验溶液呈澄明红色,管底无细胞残留或少量残留,表明有溶血反应;若细胞全部下沉,上清液无色透明,表明无溶血现象发生[14]。
1.2.3 无患子总皂苷纯度鉴定
1.2.3.1 齐墩果酸标准曲线制备 根据HPLC-MS分析结果,无患子总皂苷主要为齐墩果酸型三萜皂苷,所以选择齐墩果酸作为无患子总皂苷元的标准品。齐墩果酸用甲醇溶解,配制成1mg/mL的溶液,取18支20mL比色管,编号1~6,每个编号三组平行,1~6号试管分别取0、200、400、600、800、1000μL齐墩果酸标准溶液,水浴挥干。每管加入0.4mL 5%(wt%)香草醛-冰醋酸溶液和1.4mL高氯酸溶液为显色剂,摇匀,在70℃水浴加热20min,冰水浴中冷却3min,加入10mL冰醋酸为溶剂,摇匀,用分光光度计在400~600nm扫描光谱,并在550nm测吸光度[15]。
1.2.3.2 无患子总皂苷水解条件的确定 浓盐酸水解法是通过皂苷经水解后得到皂苷元,再经换算得到相应皂苷的含量。无患子皂苷根据文献[16]对于酸种类、酸浓度及水解时间都有一定的研究,但关于水解方式的比较方面报道甚少。本实验比较了三种不同的水解方式:回流加热、100℃水浴、高温高压。方法如下:
待测样品的配制:精密称取无患子总皂苷粉末1g,加33mL水溶解后,再加入17mL浓盐酸使盐酸浓度达到35%左右。
(a)回流加热:组装实验室常用的回流冷凝装置,将上述待测样品置于100mL圆底烧瓶中,用油浴锅在100℃回流加热2h;
(b)100℃水浴:将上述待测样品置于100mL锥形瓶中,加硅胶塞密封,用水浴锅在100℃加热2h;
(c)高温高压:将上述待测样品置于100mL锥形瓶中,加硅胶塞密封,用高压蒸汽灭菌锅在120℃,0.1MPa加热2h。
将以上三种水解方式的产物用三氯甲烷振摇提取三次,合并提取液,回收溶剂至干,甲醇定容后待测定。
1.2.3.3 无患子总皂苷与齐墩果酸量比换算系数 齐墩果酸的分子式C30H48O3,分子量为456,按含量加权分析[17]推断无患子总皂苷的平均分子量为882.5,与齐墩果酸的分子量在理论上的量比系数为1.94,考虑在无患子总皂苷水解过程中不能达到完全水解,无患子总皂苷元的得率有所损失,皂苷元最大得率达85%左右[18-19],所以量比系数1.94除以85%即为加权系数2.3,无患子总皂苷含量的计算公式为:
无患子总皂苷的含量=试样中齐墩果酸相当量(μg)×2.3
2 结果与分析
2.1 大孔树脂分离所得无患子粗皂苷液成分鉴定
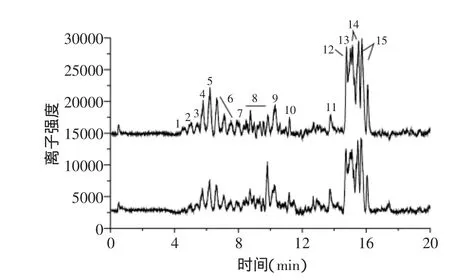
图1 无患子粗皂苷液的ESI检测总离子流色谱图Fig.1 ESI-total ion chromatograms of TSS
一般三萜皂苷类化合物的紫外吸收很弱,通过高效液相-紫外检测(HPLC-UV)或高效液相-蒸发光散射(HPLC-ESCD)测定其含量较困难,且需要较昂贵的对照品。HPLC-MS检测三萜皂苷类物质灵敏度高,并能提供每个谱峰的分子量及结构信息,在没有对照品的情况下,可以快速鉴定出三萜皂苷类成分。本研究首先采用大孔树脂初步分离纯化得无患子粗皂苷液,为了判断所得无患子粗皂苷液中的成分,选择正离子和负离子两种模式同步进行无患子粗皂苷的ESI-MS检测,正、负离子模式下无患子粗皂苷成分总离子流色谱图如图1所示,可以看出其单体成分较多,很难完全分离,对主要色谱峰进行编号,编号为1~15。
正离子ESI-MS谱中出现各皂苷成分的[M+Na]+准分子离子峰,负离子ESI-MS谱中则为[M-H]-和[M+Cl]-准离子峰。通过正、负离子质谱相互验证,因为各单体很难完全分离,所以质谱图中会有一些碎片离子干扰,但主要的碎片离子可以较易从图中读出,并在相应的碎片离子上标明[M+Na]+、[M-H]-、[M+Cl]-,从而初步判断无患子粗皂苷中主要单体的分子量,根据相关书籍[20],初步推测无患子粗皂苷液中主要成分,结果见表1。
根据质谱元素分析软件Mass Lynx V4.1,可以初步判断分子量1188、1086、1400为含氮或含硫化合物,属于杂质成分;6号组峰分子量均为1190,该类物质王晓淳等[21]报道其性质不稳定,很难分离得到纯品,结构难以鉴定;8号组峰主要是分子量为1086与1232的混合峰,很难分开;分子量为1044和1128的物质至今未见报道;分子量为1000、1002、1146、1148、1232的物质属于倍半萜糖苷类化合物;分子量为1206、1074、882、750、924、966的物质为三萜皂苷类化合物,杨运云等[22]对复方毛冬青冲剂中三萜皂苷活性成分进行分析,报道了齐墩果酸型及乌索烷型三萜皂苷在一级正离子质谱图中均有m/z437特征离子,上述无患子三萜皂苷正离子ESI-MS中均有特征碎片峰m/z437,结果与杨运云等的报道相似,进一步确定了分子量为1206、1074、882、750、924、966为三萜皂苷,且主要为齐墩果酸型三萜皂苷。结合图3和表1可知,4~10min出峰的主要是倍半萜糖苷类物质,10~20min出峰的主要为三萜皂苷类物质,且主要有8种三萜皂苷单体,这与Huang等[23]和Saxena等[24]报道的10~20min出现约7个三萜皂苷单体的报道相似。综上所述,要得到纯度较高的皂苷成分,还需要进一步分离,将倍半萜糖苷类物质去除。
2.2 无患子总皂苷进一步纯化
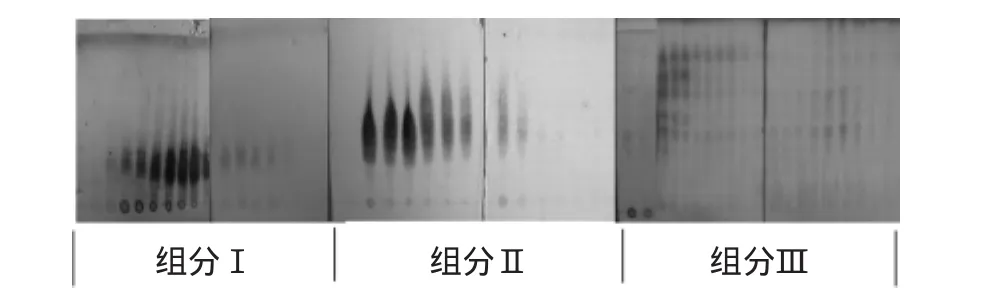
图2 TLC同步检测MCI柱分离结果Fig.2 The result of separation of MCI GEL by TLC detection
2.2.1 TLC同步监测MCI柱分离结果 根据不同物质在MCI柱上的吸附力不同,先用蒸馏水洗脱直至用薄层板检测无检出物,这部分合并即为组分Ⅰ;用30%乙醇洗脱至薄层板上无检出物,合并洗脱液成组分Ⅱ;再用70%乙醇洗脱直至无目标物,合并洗脱液得组分Ⅲ,根据洗脱物保留时间不同可知组分Ⅰ、Ⅱ、Ⅲ是三类不同极性的物质。
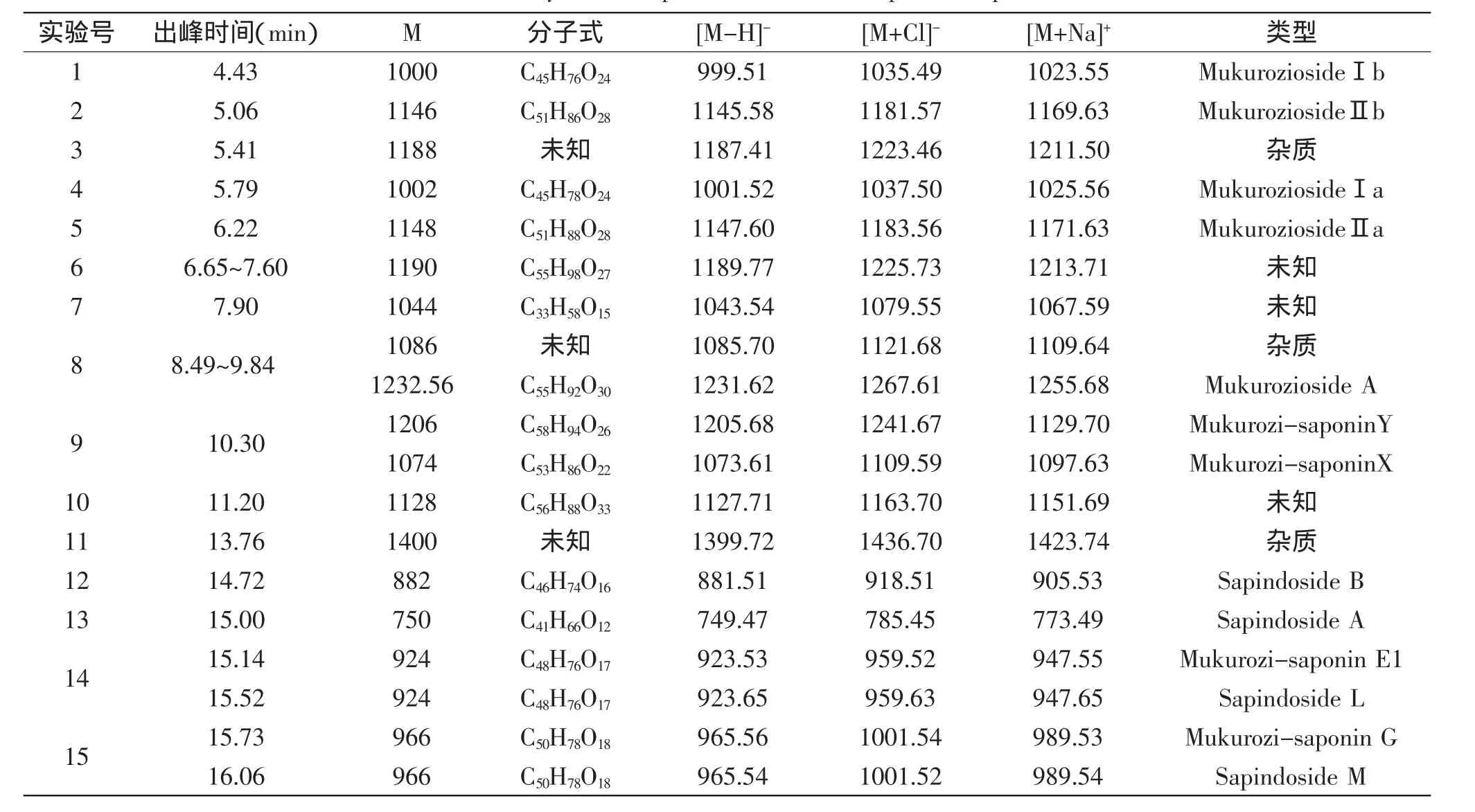
表1 无患子粗皂苷成分分析Table 1 Analysis of components of the total sapindus-saponins
2.2.2 HPLC-MS检测组分Ⅰ、Ⅱ、Ⅲ 结合表1所鉴定的无患子粗皂苷成分,编号对应图3。根据分子量及出峰时间可以推断无患子总皂苷主要集中在组分Ⅲ中,主要成分如下(括号内为分子量):Sapindoside B(882)、Sapindoside A(750)、Mukurozi-saponin E1(924)、Sapindoside L(924)、Mukurozi-saponin G(966)、Sapindoside M(966);组分Ⅱ中也含有少量无患子三萜皂苷,分别为Mukurozi-saponinY(1206)和MukurozisaponinX(1074),出峰时间为10.02min,但因为其含量较少,且从组分Ⅱ中完全分离比较困难,所以本实验未对组分Ⅱ进行进一步分离,组分Ⅲ即为本实验所需无患子总皂苷质控品,且根据峰面积可知组分Ⅲ中无患子总皂苷含量较高。
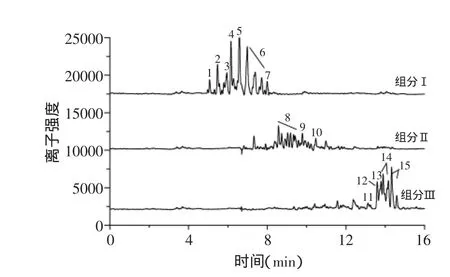
图3 组分Ⅰ,Ⅱ,Ⅲ的ESI检测负离子流色谱图Fig.3 ES-ion chromatograms of mixtureⅠ,Ⅱ,Ⅲ
2.2.3 组分Ⅰ、Ⅱ、Ⅲ性能验证 经Libermann-Buehard反应及溶血实验,发现组分Ⅰ、Ⅱ的Libermann-Buehard反应无颜色变化,在较高浓度(0.3mg/mL)仍无溶血反应,说明几乎不含皂苷或皂苷含量较少,这与HPLC-MS推测的结果一致;组分Ⅲ有明显的皂苷的特性,Libermann-Buehard反应颜色由黄-红-紫-蓝,且在较低浓度(0.02mg/mL)时仍有溶血性,说明三萜皂苷含量较高,进一步断定组分Ⅲ即所需无患子总皂苷质控品。
2.3 无患子总皂苷纯度的判定
通过MCI柱分离可知,组分Ⅲ为纯度较高的无患子总皂苷,因为其含有相同的齐墩果酸型皂苷元,结构相似,所以很难完全分离开,无法用面积归一化法对其进行纯度的计算。根据文献[25],通过将皂苷水解为皂苷元,再经过加权系数分析,可以计算皂苷的含量。
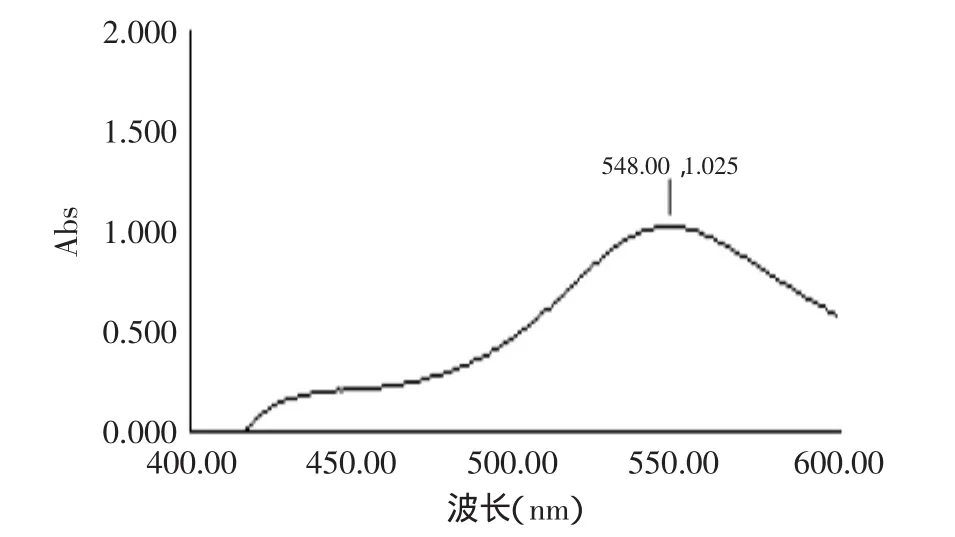
图4 齐墩果酸在香草醛-冰醋酸体系中紫外吸收Fig.4 The UV graph of olenolic acid in vanilin-acetic-HClO4system
2.3.1 齐墩果酸标准曲线 齐墩果酸通过香草醛-冰醋酸显色后在400~600nm扫描,可知在550nm左右有最大吸收。以齐墩果酸为无患子三萜皂苷元的标准品,得回归方程如下:y=0.0006x+0.0838,R2=0.9996,其中,x为齐墩果酸含量,y为吸光度。齐墩果酸含量在200~1000μg范围内与吸光度呈现良好的线性关系,可作为齐墩果酸型皂苷元质量评价的标准曲线。
2.3.2 无患子总皂苷水解后的吸收特征 无患子总皂苷经过水解得到皂苷元,经过香草醛-冰醋酸显色后,在550nm左右处有最大吸收,这与齐墩果酸标准品的最大吸收峰一致,进一步说明了该三萜皂苷元为齐墩果酸型皂苷元。
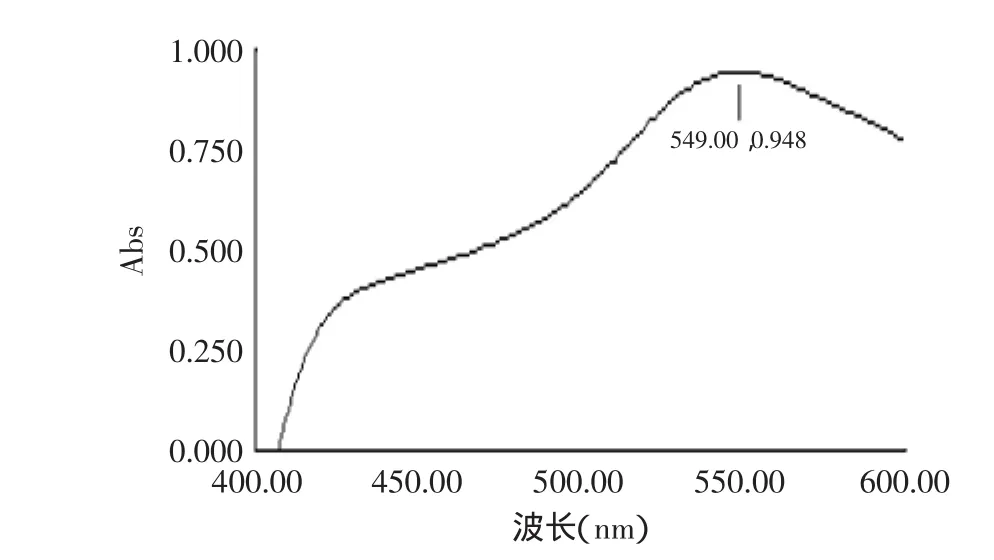
图5 无患子皂苷元在香草醛-冰醋酸体系中紫外吸收Fig.5 The UV graph of sapindus-sapogenin in vanilin-acetic acid-HClO4system
2.3.3 无患子总皂苷水解条件的确定 从表2可以看出,当盐酸浓度35%,水解2h时,高温高压方法下皂苷元得率最高,通过1.2.3.3的换算方法可知无患子总皂苷的纯度可达95.2%。
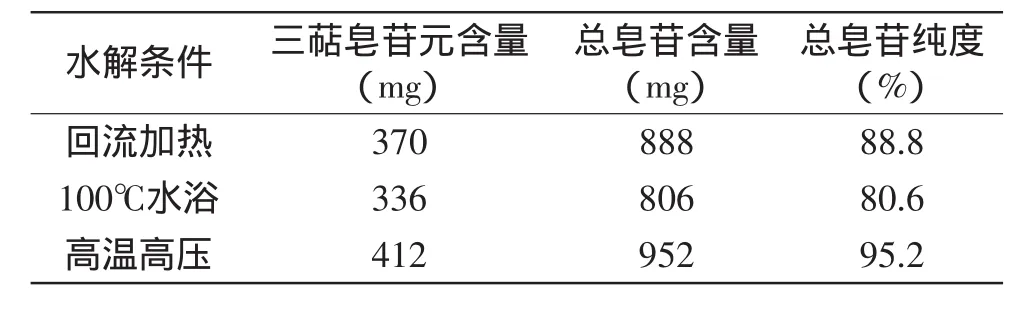
表2 无患子皂苷水解条件的确定(n=3)Table 2 Comparation of hydrolysis conditions of the total sapindus-saponins(n=3)
3 结论
通过乙醇提取,石油醚除杂,正丁醇萃取,AB-8大孔树脂,MCI凝胶柱分离纯化得无患子总皂苷,鉴定主要为齐墩果酸型三萜皂苷,纯度可达95.2%,可作为无患子总皂苷质控品。无患子皂苷是一类结构相似的混合物,由于其具有多样性与复杂性,单个皂苷分离十分困难,关于无患子皂苷的化学组成的研究一直是一大难题。本文的研究内容为进一步分离无患子皂苷单体奠定了坚实的基础。
[1]Simone CBG,Eloir PS,Valquiria LB.HPLC method to assay saponins in Ilex paraguariensis aqueous extrat[J].J Braz Chem Soc,2005,16(4):723-726.
[2]Sezgin C-AE,Artik N.Determination of saponin content in turkish tahini halvah by using HPLC[J].Advance Journal of Food Science and Technology,2010,2(2):109-115.
[3]Combarieu ED,Falzoni M,Fuzzati N,et al.Identification of ruscus steroidal saponins by HPLC-MS analysis[J].Fitoterapia,2003,73:583-596.
[4]Liu J H,Wang X.Analysis of the constituents in the Chinese drug notoginseng by liquid chromatography-electrospray mass spectomntry[J].J Chin Pham Sci,2004,13(4):225-237.
[5]Ryan D,Robards K,Prenzler P,et al.Liquid chromatography with electrospray ionization mass spectrometric detection of phenolic compounds from Olea euro paea[J].J Chromatogr A,1999,855(2):529-537.
[6]Lee MR,Chen CM,Wang BH,et al.Analysis of Saponins from black bean by rlectrospray ionization and fast atom bombardment tandem mass spectrometry[J].Mass Spectrom,1999,34:804-812.
[7]Feng L,Li JL,Zeper A,et al.Structural characterization of steroidal saponin by electrospray ionization and fast-atom bombardment tandem mass spectrometry[J].Rapid Commun Mass Spectrom,2002,16:1168-1173.
[8]Wang X,Sakuma T,Asafu-AE,et al.Determination of ginsenoside in plant extracts from panax ginseng and panax quinquefolius by LC/MS/MS[J].Anal Chem,1999,71:1579-1584.
[9]何伟,李伟.大孔树脂在中药成分分离中的应用[J].南京中医药大学学报,2005,21(2):134-136.
[10]Shiau IL,Shih TL,Wang YN,et al.Quantification for saponin from a soapberry(sapindus mukorossi gaertn)in cleaning products by a chromatographic and two colorimetric assays[J].J Fac Agr,2009,54(1):215-221.
[11]Hamburger M,Slacanin I,Hostettmann K,et al.Acetylated saponins with molluscicidal activity from Sapindus rarak:unambiguous structure determination by proton nuclear magnetic resonance and quantitative analysis[J].Phytochemical Analysis,1992,3(5):231-237.
[12]高锦明.植物化学[M].北京:科学出版社,2003,267-271.
[13]Hong XS,Yi PY,Hang JP,et al.Adjuvant effect of panax notoginseng saponins on the immune responses to ovalbumin in mice[J].Vaccine,2004,22:3882-3889.
[14]秦枫,陈玉勇,冒美丽,等.三七皂苷PNS-5的溶血性及生物免疫活性研究[J].中国医药导报,2008,5(29):10-11.
[15]Xiang ZB,Tang CH,Chen G,et al.Studies on colorimetric determination of oleanolic acid in Chinese quince[J].Natural Product Research and Development,2001,13(4):23-26.
[16]袁丁,何毓敏,鲁科明,等.Rp-HPLC法测定竹节参药材中三萜皂苷元齐墩果酸的含量[J].中国药房,2009,20(15):1151-1153.
[17]刁小琴,田晓菊,关海宁,等.以齐墩果酸为标准品测定大豆皂甙的研究[J].精油加工,2009(11):53-55.
[18]刘新清,王小艺.茶皂素含量的分析[J].茶叶,2000,26(2):81-82.
[19]钟世红,卫莹芳,古锐.红毛五加叶中常春藤皂苷元的高效液相色谱法测定[J].时珍国医国药,2010,21(1):6-7.
[20]宁正祥.食品成分分析手册[M].北京:中国轻工业出版社,1997.
[21]王晓淳.高效液相色谱-质谱联用分析无患子中的表面活性物质[J].高等学校化学学报,2006,19(6):529-531.
[22]杨运云,郭鹏然,陈怡禄,等.高效液相色谱-质谱法快速鉴定复方毛冬青冲剂中三萜皂苷活性成分[J].分析化学研究简报,2009,37(10):1523-1527.
[23]Huang HC,Liao SH,Cheang FR,et al.Molluscicidal saponins from Sapindus mukorossi,I nhibitory agents of Golden Apple Snails,Pomacea canaliculata[J].J Agr Food Chem,2003,51:4916-4919.
[24]Saxena D,Pal R,Dwivedi AK,et al.Characterisation of sapindosides in Sapindus mukorosii saponin(reetha saponin)and quantitative determination of sapindoside B[J].Journal of Scientific & Industrial Research,2004,63(2):181-186.
[25]李锐,周燕,杨永成,等.无患子皂苷成分的串联质谱分析[J].高等学校化学学报,2006,27(1):52-54.
猜你喜欢
中南林业科技大学学报(2022年1期)2022-02-23
奥秘(创新大赛)(2021年2期)2021-04-07
山东林业科技(2019年2期)2019-06-03
中成药(2018年10期)2018-10-26
天然产物研究与开发(2018年7期)2018-08-21
天然产物研究与开发(2018年5期)2018-06-13
中成药(2017年9期)2017-12-19
中成药(2017年9期)2017-12-19
天然产物研究与开发(2016年1期)2016-06-05
医学研究杂志(2015年8期)2015-06-22
