
配体保护下的Au8团簇吸附和活化O2的第一性原理研究
2013-01-04 06:09余盛萍高艳蓉黄德林杨明理
成都工业学院学报 2013年4期
余盛萍*,高艳蓉,黄德林,杨明理
(1.西南民族大学 化学与环境保护工程学院,成都 610041;2.四川大学 原子与分子物理研究所,成都 610065)
1987年Haruta等[1-2]发现TiO2或Fe2O3载体上的Au团簇在低温下对CO的氧化反应有良好的催化作用,打破了Au([Xe]5d106s1)没有催化活性的传统观念,在世界范围内掀起了研究Au团簇的热潮。Au团簇在低温下可以催化一系列反应,如CO的氧化[3]、H2O2的形成[4]、丙烯的环氧化[5]等。研究表明尺寸较小的Au团簇具有较高催化活性,但也使其反应条件更加苛刻。粒径小的Au团簇容易聚集成大团簇,以降低表面能,使团簇更稳定,但导致其催化活性降低。为了阻止小团簇之间的聚集,近年来采取了有机配体保护团簇的方法。这些配体包括PH3[6]、硫醇盐[7]、炔烃[8]等。最近,Tsunoyama 等[9]发现配体 poly(N-vinyl-2-pyrrolidone)(PVP)能够保护Au团簇且不降低其催化活性。Aun∶PVP复合物对于O2参与的系列反应显示出很高的反应活性和选择性[10],但其催化活化机理至今未被明确阐述。最近,笔者在TPSS/def2-TZVP水平下研究了Au32团簇与单个PVP之间的相互作用,找到了它们之间的作用模式,揭示了单个PVP保护下Au32能够保持活性的原因[11]。在此基础上,本文利用密度泛函理论(DFT)继续研究了Au8与单个或多个PVP之间形成吸附产物的几何结构和电子结构,探讨PVP与Au8团簇之间的作用机理,为研究配体保护下Aun的催化反应机理奠定了基础。
1 计算方法
为了简化计算,用H代替PVP中的-(CH2-CHR)n基团。为了与PVP区别,配体简称PRD。Au8表面有多个可能的吸附位点,Au8与PRD之间有多个可能的吸附方式。因此,我们构建了大量Au8∶PRDn(n=1~4)起始结构,在TPSS[12]/def2-QZVPP水平下对Au8-PRDn(n=1~4)的起始构型进行优化和遴选,得到较为稳定的吸附产物。在此基础上,采用类似方式设计O2的吸附方式和初始构型,经过再次构型优化和遴选,得到系列较稳定的PRD-Au8-O2构型,并在相同水平下计算了所得构型的振动频率,其频率全为正,以确定其为稳定构型。为了测试泛函的适用性,我们同时用另2种泛函PBE[13]和BLYP[14]进行了类似计算,发现这2种泛函得到的结果与TPSS的结果基本一致。
所有计算采用Turbomole程序包完成。采用了Wood-Boring的相对论校正(MWB)[15]有效核电势来描述Au的60内层电子,针对5s、5p、5d和6s价层电子采用了4重分裂的价电子基组加上扩展的极化函数,对C、N、O和H等也采用def2-QZVPP基组。计算得到O2和Au8中的O-O和Au-Au键长分别是1.22和2.61,与实验值 1.21和 2.67结果相近。
2 计算结果与讨论
2.1 Au8团簇与PRD之间的相互作用
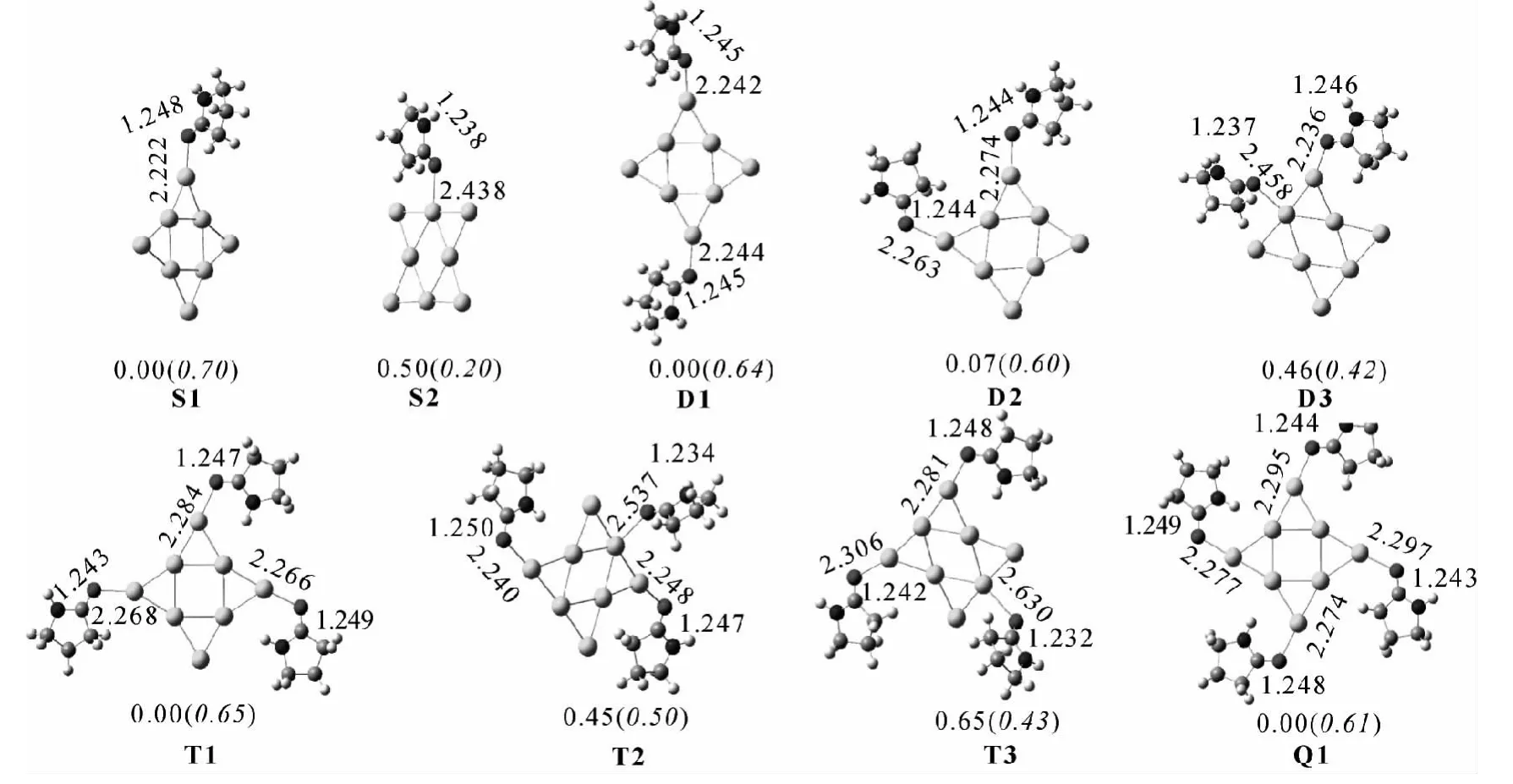
图1 Au8∶PRDn(n=1~4)的较稳定异构体的结构,键长,只标出 Au 和 C键长。结构下的数据为相对能量/eV,括号里是平均吸附能/eV
图1列出了优化得到较为稳定的吸附产物Au8∶PRDn(n=1~4)构型。命名中的字母S、D、T、Q分别表示Au8吸附1,2,3和4个PRD,数字表示异构体的能量顺序。例如D2代表由2个PRD吸附到Au8得到的次稳定结构Au8∶PRD2。在图1所示所有构型中,吸附都发生在PRD中的O原子与Au8中的单个Au原子之间。我们计算了其他多种吸附方式,包括桥位和多位等,但得到的附产物能量明显高于图1中的单吸附方式。Au8∶PRD只有2种异构体,PRD吸附到Au8的顶点得到最稳定的吸附产物S1,比S2稳定0.50 eV。Au8∶PRD2有邻位、间位和对位3种异构体,其中对位异构体D1最稳定,其能量比D2和D3的分别低0.07 eV和0.46 eV。在Au8∶PRD3异构体中,3个PRD都吸附在顶点的T1最稳定,其能量分别比T2和T3低0.07 eV和0.46 eV。Au8∶PRD4仅有1种较为稳定的吸附产物Q1,4个PRD都吸附在顶点位置。比较Au8和Au8∶PRDn(n=1~4),吸附PRD后Au8的构型基本不变,只是吸附位点附近的Au-Au键长发生少许改变,且距离吸附位点愈远,改变越小。形成的Au-O键长在2.132到 2.478之间变化,而PVP上C-O键在1.245与 1.261之间变化。
我们定义PRD在Au8上的平均吸附能为:这里的 EPRD、EAu8和 EAu8∶PRDn分别表示PRD,Au8,和Au8∶PRDn的能量。图1中各结构下方给出了它们的平均吸附能数值。最稳定 Au8∶PRDn(n=1~4)异构体S1、D1、T1和Q1的平均吸附能在0.61~0.70 eV之间变化,且随着PRD数目增加,Eads有减小的趋势,但减小量较小。
2.2 O2和 Au8∶PRDn(n=1 ~3)上的吸附
由于Au8∶PRD4中Au8的4个顶点都被PRD占据,O2无法再进攻其余位点并形成稳定吸附产物。因此,我们以Au8∶PRDn(n=1~3)的构型为基础,进一步研究它们对O2的吸附,探索可能存在于PRD和O2在Au8上的共吸附效应,并对PRD保护下Au8保持活化O2的能力的原因进行分析。我们计算了所有吸附位置对应的吸附产物结构,由于其他构型的能量相对较高,稳定性较差。图2只列出了经过结构筛选和优化得到的最稳定的Au8-PRDn-O2(n=1~3)构型,同时给出了Au8-O2的最稳定构型以便比较。这3个含PRD的结构中的Au-O键长变化范围为 2.038 ~ 2.171,略短于不含PRD的 Au8-O2中的 Au-O键长(2.178。相应地,O-O键长变化范围为1.254~1.304略长于Au8-O2中的O-O键(1.251同时,O-O键长比自由O2分子的O-O键长(1.219长。这说明配体PRD的存在加强了O2与Au的作用,并有助于活化O2。随着PRD配体数目的增加,这种增强作用更为显著。
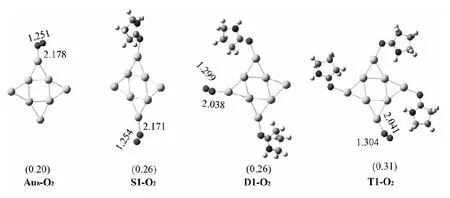
图2 基态Au8-O2和Au8-PRDn-O2(n=1~3)的分子构型,键长只标出Au-O和O-O键长括号内是吸附能/eV。
类似情形,O2在 Au8∶PRDn上的定义吸附能为 Eads=EAu8∶PRDn+EO2- EAu8∶PRDn∶O2,其中 EAu8∶PRDn、EO2和EAu8∶PRDn∶O2分别表示 Au8∶PRDn、O2和 Au8∶PRDn∶O2的能量。如图1 中括号里的吸附能所示,O2在有配体PRD保护的Au8团簇Au8∶PRDn(n=1~3)上的吸附能比无配体的Au8团簇的吸附能分别大0.06、0.06和0.11 eV。吸附能数据进一步表明,保护配体PRD有助于O2的吸附,且随着PRD数目的增多,吸附能有增大的趋势。
2.3 O2和PRD在Au8团簇上吸附产物的电子性质
对 PRD吸附产物 Au8∶PRDn(n=1~4)的NBO分析显示,Au8的电荷总数变化范围为 -0.10 ~ -0.25 e,顶点的 Au 为负电,其他的Au为正电。有部分电子从PRD转移到Au8,但总电荷转移量比较小。可见,PRD的作用不仅仅限于吸附在Au8表面阻碍其他小团簇的靠近,而且部分地改变了Au8的电子结构,会对其活性产生部分影响。表1列出了在TPSS/def2-QZVPP水平下最稳定的 Au8∶PRDn∶O2(n=1 ~3)构型中各原子的净电荷和自旋密度。分析O2,PRD和Au83部分的净电荷变化,可以发现非常重要的电荷转移规律。在Au8∶PRDn∶O2(n=1~3)中,PRD和Au8的净电荷为正,O2的净电荷为负,说明电子从PRD及Au8流向O2,转移到O2部分的净电荷在0.30 e左右。Au8有2方面的作用:1)电子供体,提供电子给O2;2)电子转移桥梁,PRD的电子经由Au8到达O2。PRD的作用值得关注,由于提供电子给Au8,导致Au8的亲电性增加,有利于与具有强吸电子性的O2的作用。由此可见,PRD与O2之间存在协同效应,共吸附左右下更有利于彼此的吸附。NBO分析显示大部分的自旋密度集中于O2部分,自旋总数约1.6 e,其余部分自旋主要分布于Au8上,在PRD上的自旋密度极小,表明O2在Au8∶PRDn上吸附后被部分活化,但O2仍然保持了O2分子的主要特征和性质,与Au8之间的作用本质上是分子吸附,不是解离吸附。
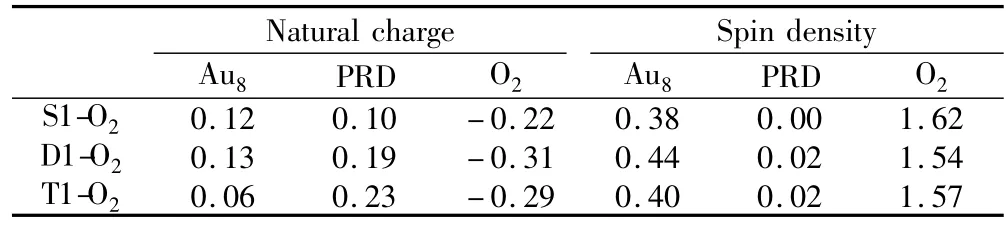
表1 Au8:PRDn:O2(n=1~3)的基态结构的净电荷和自旋密度分布
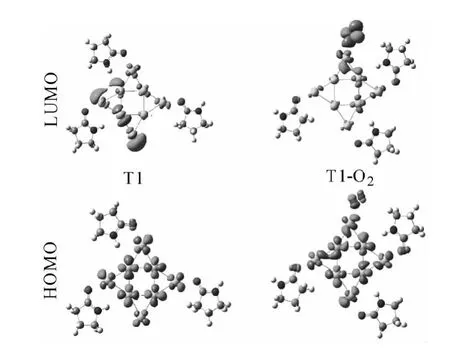
图3 Au8∶PRD3 和 PRD3:Au8∶O2基态结构的HOMO和LUMO轨道
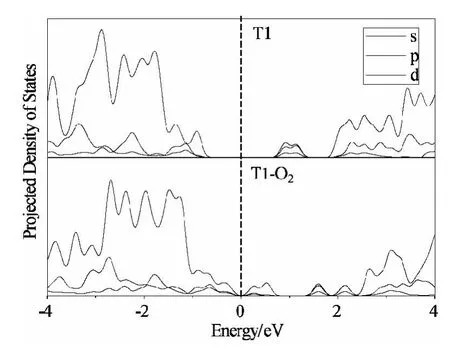
图4 Au8∶PRD3和Au8∶PRD3:O2的基态结构的PDOS
我们进一步分析Au8∶PRDn和PRDn∶Au8∶O2(n=1~3)的基态结构的最高占据分子轨道(HOMO)和最低空轨道(LUMO)的构成。如图3所示,Au8∶PRD3的HOMO和LUMO主要分布在Au8团簇上,仅有极少量分布在与Au相连的O原子上。由于Au8在吸附作用中提供电子,主要是其HOMO做出贡献。因此O2可以被吸附到Au8中空间许可的位置。Au8中无配体保护的顶点位置最有利于O2的吸附。因此得到的吸附产物Au8∶PRDn∶O2中O2都吸附在顶点位置上。另一方面,吸附作用发生后,LUMO主要分布在被吸附且活化了的O2上,表面吸附产物可以参与氧化反应,且反应的活性部位位于O2。图4显示了Au8∶PRD3和Au8∶PRD3∶O2基态结构的态密度(PDOS)。Au8∶PRDn和Au8∶PRDn∶O2的HOMO和LUMO 能隙相比较,前者能隙为1.820 eV,明显比后者(0.522 eV)大,表明发生O2吸附后,吸附产物具有更高的化学活性。Fermi能级附近的占据轨道都由s、d和p等成分组成,但吸附后d成分更靠近Fermi能级,表明O2吸附导致Au中d电子的活性提高。同时,O2吸附让空轨道更靠近Fermi能级并改变了空轨道的组成,由吸附前的主要由s和p成分贡献,转变为主要由p成分贡献,表明空轨道的活性增加并以p轨道参与氧化反应。
3 结语
在TPSS/def2-QZVPP水平下,分析了配体PRD对Au8团簇的保护机制,探讨了一系列Au8与1~4个PRD之间的相互作用及PRD保护下Au8(n=1~4)对O2的吸附和活化作用,得到以下结论:1)PRD倾向于吸附在Au8团簇的4个顶点上,PRD中O与Au间形成较弱的Au-O键,有少量的电子从PRD转移到Au8,从而部分改变了Au8的电子结构;2)PRD和O2在Au8团簇上发生了协同吸附效应,PRD使Au8团簇催化活性更强,且随PRD的个数增加对O2的吸附和活化作用增强;3)PRD和O2在Au8团簇上的协同吸附效应的本质是电荷转移机制,PRD提供电子给Au8,加强了Au8的亲电性,有利于O2的吸附和活化;Au8既是电子供体,又是电子传递的桥梁。
[1]HARUTA M,KOBAYASHI T,SANO H,et al.Novel gold catalysts for the oxidation of carbon monox ide at a temperature far below 0 ℃ [J].Chem Lett,1987,16(2):405 -408.
[2]HARUTA M,YAMADA N,KOBAYASHI T,et al.Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide[J].J Catal.,1989,115(2):301 -309.
[3]XU C X,SU J X,XU X H.Low temperature COoxidation over unsupported nanoprous gold[J].J.Am.Chem.Soc.,2007,129(1):42 -43.
[4]SIVADINARAYANA C,CHOUDHARY T V,DAEMEN L L.The nature of the surface species formed on Au/TiO2during the reaction of H2and O2:An inelastic neutron scattering study[J].J.Am.Chem.Soc.,2004,126(1):38 -39.
[5]IDAKIEV V,TABAKOVA T,NAYDENOV A,et al.Gold catalysts supported on mesoporous zirconia for low-temperature water-gas shift reaction[J].Appl.Catal.B:Environ.,2006,63(3 -4):178 -186.
[6]WALTER M,MOSELER M,WHETTEN R L.The halogen analogs of thiolated gold nanoclusters[J].Chem.Sci.,2011,2(13):1583 -1587.
[7]NEGISHI Y,NOBUSADA K,TSUKUDA T.Origin of magic stability of thiolated gold clusters:a case study on Au25(SC6H13)18 [J].J.Am.Chem.Soc.,2005,127(14):5 261 -5 270.
[8]MAITY P,TSUNOYAMA H,YAMAUCHI M.Stabilized gold clusters:from isolation toward controlled synthesis[J].J.Am.Chem.Soc.,2011,133(50):20 123-20 125.
[9]TSUNOYAMA H,ICHIKUNI N,SAKURAI H.Effect of electronic structures of Au clusters stabilized by poly(N-vinyl-2-pyrroildone)on aerobic oxidation catailsis[J].J.Am.Chem.Soc.,2009,131(20):7 086 -7 093.
[10]ZHANG H J,OKUNURA M,TOSHIMA N.Stable dispersions of PVP-Protected Au/Pt/Ag trimetallic nanoparticles as highly active colloida l catalysts for aerobic glucose oxidation[J].J.Phys.Chem.C,2011,115(30):14 883 -14 891.
[11]YU S P,ZENG Q,LOU Z Y,et al.First-principles study of O2activation on ligand-protected Au32clusters[J].Phys.Chem.Chem.Phys.,2013(15):9 742-9 751.
[12]TAO J,PERDEW J P,STAROVEROV V N,et al.Climbing the density dunctional ladder:nonempirical meta-generalized gradient approximation designed molecules and solids[J].Phys.Rev.Lett,2003,91(14):1 -4.
[13]PERDEW J P,BURKE K,EMZERHOF M.Generalized gradient approximation made simple[J].Phys.Rev.Lett.,1996,77(18):3 865 -3 868.
[14]BECKE A D.Density-functional exchange-energy approximation with correct asymptotic behavior[J].Phys.Rev.A,1988,38(6):3 098 -3 100.
[15]WOOD J H,BORING A M.Improved Pauli Hamiltonian for local-potential problems[J].Phys.Rev.B,1978,18(6):2 701 - 2 706.
[16]ANGELA N S,DAVID T M.Charge-Dependent trends in strucres and vibrational frequencies of[CO-Au-O2]q(q= -1,0,+1)complexes:evidence for cooperative interactions[J].J.Phys.Chem.A,2012,116(37):9 370 -9 381.
[17]JIANG H L.Au@ZIF-8:CO oxidation over gold nanoparticles deposited to metal-Organic framework[J].J.Am.Chem.Soc.,2009,131(32):11 302-11 303.
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
数学物理学报(2022年3期)2022-05-25
浙江化工(2022年4期)2022-05-07
数学物理学报(2022年1期)2022-03-16
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年3期)2021-07-19
青岛大学学报(工程技术版)(2019年2期)2019-09-10
国外医药(抗生素分册)(2016年4期)2016-07-12
信息记录材料(2016年4期)2016-03-11
枣庄学院学报(2015年5期)2016-01-09