
厌氧微生物作用下土壤中砷的形态转化及其分配
2013-01-03 06:05:04杨明许丽英宋雨王新贾永锋
生态毒理学报 2013年2期
杨明,许丽英,宋雨,王新,贾永锋
中国科学院沈阳应用生态研究所中国科学院污染生态与环境工程重点实验室,沈阳110016
我国土壤重金属污染问题日益严重,与砷相关的土壤健康与环境安全问题逐渐受到广泛关注。由于天然原因和矿冶行业发展等因素,目前我国南方地区如湖南、广东、贵州和云南的某些地区土壤砷污染较为严重。如湖南郴州由于采矿和冶炼导致大面积农作物砷含量超标,所出产的多种蔬菜砷含量达到7.9 ~16.6 mg·kg-1[1],云南某旧矿区韭菜中砷含量甚至高达89.02 mg·kg-1[2],砷含量超标的农产物可直接对人和禽畜造成毒害。同时,土壤中的砷可经由生物与化学因素的作用发生转化或迁移,进而间接污染地下水。因此,土壤中砷的赋存形态变化与迁移转化等环境行为成为学者广泛关注的科学问题。
土壤砷污染的环境效应与砷在土壤中的赋存形态及其迁移转化过程密切相关。环境中的砷主要以+3 价的亚砷酸盐和+5 价的砷酸盐形式存在[3]。由于价态的不同,As(Ⅲ)和As(Ⅴ)的毒性和迁移性也存在差异。土壤中的砷主要与黏土矿物、铁、铝、锰氧化物矿物和有机质等结合,厌氧微生物的活动强烈地影响土壤中砷的赋存形态及其地球化学过程。微生物对砷的直接还原作用以及对相应固砷矿物的转化都可引起砷的活化或释放[4-8],同时也会对砷在土壤中的结合形态产生影响,进而影响砷的生物有效性、环境行为及其在不同形态间的再分配。不少学者研究发现,土壤中砷的环境行为是由微生物对铁和砷的还原作用调控的[9-11]。而厌氧条件下微生物还原作用亦可同步驱动硫元素的还原过程,砷与铁的还原及后续发生的硫酸盐还原过程相互交错。硫酸盐还原产生的S(-Ⅱ)与铁还原产生的Fe(Ⅱ)可反应形成各种铁硫矿物(硫化亚铁、砷黄铁矿和黄铁矿等),在次生矿物沉淀的过程中游离态的砷可能经由吸附或共沉淀作用被固持[12-13]。因此,厌氧微生物作用下土壤中砷的赋存形态也变得相当复杂,目前针对微生物作用下土壤中As、Fe、S 还原过程中,砷的形态转化与相应铁元素形态变化的动态过程,以及当发生硫酸盐还原反应时,硫离子参与的砷、铁二次固定过程中砷的赋存形态仍需进行系统深入的研究。
本研究主要利用清洁土壤负载砷,模拟土壤砷污染环境,采用不同的提取方法研究厌氧微生物作用下土壤中砷-铁-硫的形态转化及其在土壤矿物中的再分配过程,考察微生物驱动的砷、铁和硫的还原过程中砷的还原与释放过程,探讨影响砷环境行为及归趋的生物-化学机制,为土壤砷污染修复提供重要依据。
1 材料与方法(Materials and methods)
1.1 土壤砷的负载
土壤样品采自沈阳张士污灌区0 ~20 cm 表层土,研磨过60 目筛。土壤消解后测得其砷、铁的本底值为分别为10.7 和34 187.5 mg·kg-1。以1∶10 的固液比进行砷的负载(液体为7.2130 mg·L-1的Na3AsO4溶液),恒温摇床35℃、170 r·min-1,避光平衡24 h。4 800 r·min-1离心10 min,倾去上清液,用蒸馏水震荡离心洗涤3 次固体,测液相中砷的残留浓度,保存固体。负载后,土壤中总砷含量为82.4 mg·kg-1。
1.2 厌氧培养实验
配制基础盐培养基(加硫体系)(MSM,g·L-1):KH2PO4,0.14;NH4Cl,0.25;KCl,0.5;CaCl2·2H2O,0.13;NaCl,1.0;MgCl2·6H2O,0.62;酵母提取物0.5;乳酸钠2 mL·L-1;外加硫源,无水Na2SO4,10 mmol·L-1。用1 mol·L-1NaOH 调节pH 为7.0 ~7.2。
培养基采用无氧水配制,配制过程中采用氮气保护以保证厌氧。在厌氧手套箱中,以1∶20 的固液比将培养基和载砷土壤加入到厌氧瓶中。非生物对照组加入培养基后121℃高压灭菌20 min。将生物组和非生物对照组样品放入摇床,35℃、170 r·min-1条件下恒温避光培养,按一定的培养时间进行取样。
取样过程在厌氧手套箱中进行,样品摇匀后取5 mL 培养液,0.22 μm 滤膜过滤。液相用于分析溶解态As(Ⅲ)、As(Ⅴ)、Fe(Ⅱ)和Fe(T)(总Fe)的浓度。滤后固体于4℃保存,用于后续盐酸提取、磷酸盐提取和酸挥发性硫化物的提取实验。
1.3 盐酸提取和磷酸盐提取
将滤过固体置于厌氧瓶中加入1 mol·L-1HCl,170 r·min-1震荡提取2 h,0.22 μm 滤膜过滤,滤液用于分析HCl 提取态As(Ⅲ)、As(Ⅴ)、Fe(Ⅱ)和Fe(T)含量。磷酸盐提取态的砷采用1 mol·L-1KH2PO4/KOH(pH5),35℃、170 r·min-1下恒温振荡提取18 h,0.22 μm 滤膜过滤,滤液用于分析磷酸盐提取态的As(Ⅲ)和As(Ⅴ)含量。
1.4 酸挥发性硫化物的提取
酸挥发性硫化物采用1 mol·L-1HCl 提取[14]。将用0.22 μm 滤膜过滤后的固体放入配有带孔胶塞的玻璃管中,加入1 mol·L-1HCl 提取1 h。氮气吹扫(氮气流速约为400 mL·min-1),0.3 mol·L-1乙酸锌和0.12 mol·L-1乙酸钠混合液作为吸收液,采用三级吸收。吸收液用于测定酸挥发性硫化物含量。
1.5 分析方法
砷的形态测定[15]:总砷As(T)和As(Ⅲ)浓度采用原子荧光光谱仪(AFS-2202,北京海光公司)测定。其中,As(T)用体积分数为10%的预还原剂进行还原(5%硫脲和5%抗坏血酸混合液),用盐酸定容,反应约10 h 后测定。As(Ⅲ)用0.4 mol·L-1的柠檬酸钠-柠檬酸缓冲液(pH4.5)作为样品稀释液和载液,于4℃保存,48 h 内测定。测定时,As(T)以盐酸为载液,As(Ⅲ)以柠檬酸钠-柠檬酸缓冲液为载液,含20 g·L-1KBH4和3 g·L-1NaOH的溶液作为还原剂进行测定。
总铁Fe(T)采用原子吸收光谱法测定,Fe(Ⅱ)采用邻菲罗啉分光光度法于波长510 nm 处测定[16]。酸挥发性硫化物含量(AVS)采用亚甲基兰比色法测定[14]。
2 结果(Results)
2.1 微生物作用下砷的释放
在微生物作用下土壤固持的砷发生了明显的还原、释放及再固定过程。厌氧培养初期,在砷还原微生物作用下,土壤吸附的砷迅速发生还原和释放(图1)。当培养实验进行到24 h 时,砷的释放量达到最大值,液相As(T)和As(Ⅲ)浓度分别为16.9 和15.5 μmol·L-1,其中As(Ⅲ)成为液相积累的砷的主要形态,约占液相总砷的91%以上。培养24 h 后液相积累的砷浓度迅速降低,液相中的砷几乎全部被再次固定到固相中,培养终点时液相残留的As(III)浓度仅为1.5 μmol·L-1。
非生物对照组实验中也有部分砷溶出,但大多以As(V)形式存在,几乎没有As(III)释放,这可能由于培养基中的乳酸盐对砷具有一定的解吸附作用造成土壤负载砷的少量溶出[17]。尽管如此,微生物还原作用仍是导致砷在不同形态间转化的根本因素。

图1 液相中砷浓度随时间的变化(ck:非生物对照组,下同)Fig.1 Variation of dissolved As concentration with incubation time(ck:abiotic control)
2.2 磷酸盐提取态砷的形态分析
利用不同提取剂对土壤中不同形态的砷进行提取,可以直接反应砷在固相中分配的变化。采用磷酸盐提取土壤中以吸附态形式存在的砷[14],研究微生物作用下吸附态砷在土壤中的形态转化,以及微生物作用下不同形态砷在土壤中的动态分配。实验中非生物对照组吸附态的As(Ⅴ)占土壤负载砷总量的48.8%左右。与之相比,在微生物还原过程中,生物组土壤中吸附态As(T)的比例呈现先降低后升高随后再降低的过程。在培养的48 h 内,吸附态的砷从45.3%降低到36.6%随后迅速增加到42.0%(图2A,表1)。这是由于在培养的24 h 内,微生物的活动造成土壤负载的砷被迅速释放进入液相,此时溶解态As(T)达到最大值(图1),在这个过程中吸附态的砷明显降低。随后被释放的砷发生再吸附过程,并且这个过程一直延续到培养的48 h 左右(图2A,表1)。厌氧培养48 h 后微生物组中磷酸盐可提取的总砷量呈降低趋势(图2A),从培养48 h 积累的0.50 mmol·kg-1下降到0.20 mmol·kg-1左右。培养结束时,微生物培养组中吸附态的砷相当于非生物对照组的44.1%。

图2 土壤中磷酸盐提取态砷浓度随时间的变化Fig.2 Variation of concentration of phosphateextracted As in soil with incubation time
与非生物对照组相比,吸附态的As(Ⅲ)含量在培养开始阶段迅速升高48 h 时达到最大值,随后稳定在左右(图2B)。而液相中的As(Ⅲ)含量在24 h 出现峰值,随后迅速降低。可见,液相中As(Ⅲ)的去除,除了土壤矿物的沉淀、共沉淀作用,固相对As(Ⅲ)的再吸附作用也具有一定贡献。

表1 厌氧微生物作用下体系中砷在固液两相的分配比例Table 1 Distribution ratio of As in solid and aqueous phase under impact of anaerobic microbial activities
2.3 盐酸提取态砷的形态分析
土壤中的铁氧化物/氢氧化物是土壤固砷矿物中的重要组成部分。厌氧微生物的作用下,土壤矿物中的铁很容易发生还原性溶解作用,其所固持的砷将发生二次释放,同时砷的不同形态之间将发生相互转化。利用不同提取剂提取可以表征相应形态砷的转化和再分配过程。盐酸可提取态砷的形态主要包括盐酸可溶解的无定形铁锰氧化物、部分结晶度很差的铁氧化物结合的砷,酸挥发性硫化物(AVS)吸附或共沉淀的砷[14]。本研究中非生物对照组土壤所负载的As(Ⅴ)总量中盐酸可提取态的砷约占34%,其余66%为盐酸不可提取的形态。生物培养过程中盐酸可提取形态的比例随着培养实验的进行呈降低趋势(图3A,表1)。培养7 d(168 h)后土壤负载的砷大约有90%以上盐酸无法提取,土壤中盐酸可提取的总砷量从逐渐降低到0.050 mmol·kg-1。

图3 土壤中盐酸提取态砷随时间的变化Fig.3 Variation of concentration of HCl-extracted As in soil with incubation time
在本研究中厌氧微生物作用下,固相中也明显观察到微生物对砷的还原作用,盐酸提取态As(Ⅲ)含量在培养48 h 内显著增加,固相中盐酸可提取的亚砷酸盐积累的最大值约为0.075 mmol·kg-1,随后的培养过程中,固相的砷发生再分配,盐酸可提取的As(Ⅲ)逐渐降低,在培养结束时盐酸提取态的As(Ⅲ)仅为而盐酸提取态As(Ⅴ)含量随时间变化一直呈现降低趋势(图3B)。这一过程表明固相中,盐酸可提取的砷酸盐部分被还原为亚砷酸盐后再次转化为其他盐酸无法提取的形态。
2.4 微生物作用下溶解态铁及盐酸提取态铁的分析
微生物驱动砷的形态转化与再分配过程中伴随着强烈的铁的还原性溶解过程(图4A 和4B)。培养实验初期,液相溶解态Fe(Ⅱ)含量显著增加,培养96 h 时液相积累的Fe(Ⅱ)含量达到此后,亚铁离子迅速降低。而非生物对照组中液相积累的Fe(Ⅱ)没有明显变化。
微生物还原作用下固相中盐酸可提取的Fe(T)比例和Fe(II)的比例也发生显著变化。利用盐酸提取土壤固相中的Fe(T)和Fe(II),研究固相中固砷矿物的结构变化及铁的再分配过程。图4B 的数据表明微生物作用造成了土壤固相中铁的活化,微生物作用下盐酸提取态Fe(T)的比例大大增加,培养168 h 后盐酸可提取的Fe(T)达到约357.1 mmol·kg-1,约占土壤中总铁的58.5%,与非生物对照相比增加了30.1%以上。固相中微生物还原产生Fe(Ⅱ)含量也随着培养实验的进行,开始阶段迅速上升,168 h 后盐酸提取态Fe(Ⅱ)含量达到125.0 mmol·kg-1。可见还原生成的Fe(Ⅱ)只有很少一部分释放到液相中,大部分仍保留在固相中。
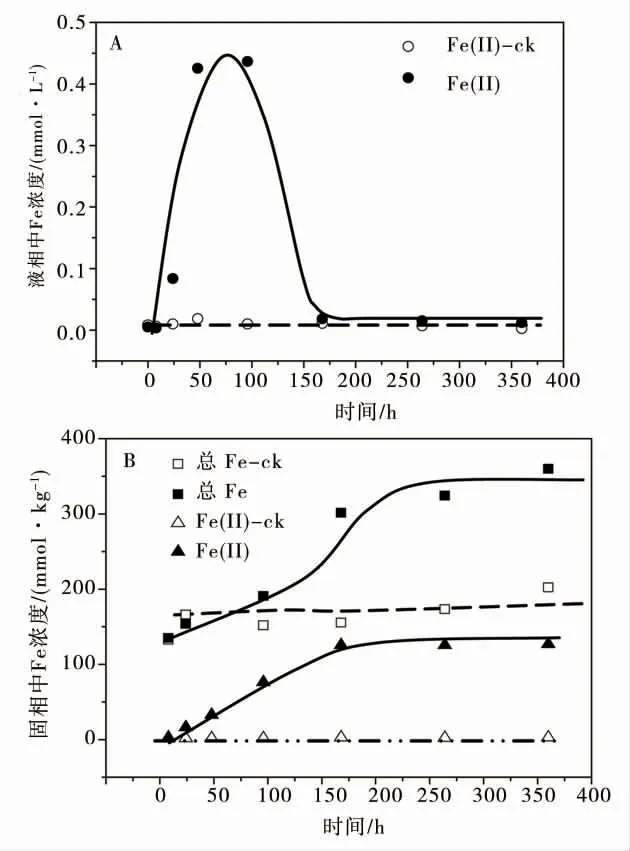
图4 液相和盐酸提取态铁浓度随时间的变化Fig.4 Variation of concentration of dissolved Fe(A)and HCl-extracted Fe(B)with incubation time
2.5 微生物作用下酸挥发性硫化物(AVS)的分析
硫酸盐微生物还原作用下产生硫离子可与体系中As(III)和Fe(Ⅱ)等离子结合而沉淀,因此硫酸盐还原过程将显著影响土壤中砷和铁再分配过程。如图5 所示,在培养实验前期,生物组中酸挥发性硫化物(AVS)含量与对照组接近。培养进行至48 h 时固相中酸挥发性硫化物含量开始迅速增加,培养96 h已观察到培养物颜色明显变黑,当培养168 h 后,酸挥发性硫化物的含量已经达到从培养96 h 开始液相Fe(Ⅱ)迅速降低(图4A),盐酸提取态Fe(Ⅱ)含量迅速增加(图4B),这一现象表明土壤固相中形成了铁硫化合物,微生物还原所产生大量的Fe(Ⅱ)形成FeS 从而被固定。

图5 酸挥发性硫化物(AVS)浓度随时间的变化Fig.5 Variation of AVS concentration with incubation time
3 讨论(Discussion)
3.1 微生物作用下土壤中砷的迁移
由于砷污染环境介质的不同、矿物组成及微生物类群等差异,砷在土壤、沉积物等复杂环境中的环境行为与归趋受到多种因素的综合影响[4,7,11,18],其中微生物还原作用也是导致砷活化进而释放的主要因素[19-20]。一般认为,铁的还原性溶解作用及直接的砷的微生物还原作用可导致铁(氢)氧化物矿物所固定的砷发生活化[4,21]。在土壤等复杂环境介质中,微生物还原作用可以同时驱动砷、铁和硫等多种元素的形态转化,土壤矿物组成极其复杂,因此砷形态转化与迁移过程与次生矿物组成与形态密切相关[18]。
由图1 和图2 的数据可知,在培养48 h 内微生物还原作用下土壤负载的砷发生了迅速的还原释放和再固定过程,As(Ⅲ)为液相积累砷的主要形态。从培养24 h 液相积累砷的最大量到培养48 h 后液相砷浓度下降到最低,液相减少的砷主要为As(Ⅲ),其总量相当于土壤负载砷总量的23.8%。培养初期吸附态As(III)保持增加,总增加量当于土壤负载砷总量的15.7%。其中培养24 h 内吸附态As(III)和释放的As(III)的增加保持同步,说明砷还原释放过程中产生的As(III)已经发生吸附(约占负载总砷的9.5%),而培养24 h 后液相As(III)迅速降低过程中吸附态As(III)仍保持增加,液相较少的As(III)中也有约6.2%的As(III)被矿物二次吸附,另外,17.6%的溶解态As(III)转化为其他形态。因此固相对砷的再吸附作用是液相中砷含量降低的原因之一。
吸附态的总砷在培养过程中也发生了迁移转化。培养前的吸附态砷总量(负载砷形态为As(V))约占负载砷总量的45.3%(表1)到培养结束时吸附态的砷总量降低到22.0%。其中约有15.7%的砷为As(III),约有5.3%的As(V)。可见80%以上吸附态的砷被转化为其他形态。采用盐酸提取土壤中铁氧化物类矿物固持的砷(图3)可看出生物培养组中盐酸可提取态砷的比例也随着培养实验的进行呈降低趋势。培养7 d(168 h)后土壤负载的砷大约有90%以上盐酸无法提取。这一过程表明,固相中的盐酸可提取的砷酸盐部分被还原为亚砷酸盐后再次转化为其他盐酸无法提取的形态。这一部分可能转化为与结晶的铁氧化物结合的砷,硫化物矿物沉淀或共沉淀的砷,如砷的硫化物矿物雄黄、雌黄等,或者铁硫共还原过程中次生黄铁矿结合的砷等这类更加稳定的赋存形态。
3.2 微生物作用下土壤中铁形态转化与砷再分配的关联
土壤中含铁矿物(如铁氧化物/氢氧化物)对砷的固持具有重要的作用,因此微生物对铁的还原作用以及由铁的还原导致的载砷矿物赋存形态与组成结构的变化都将严重影响砷在土壤中的再分配过程。在厌氧培养实验中,与非生物对照组相比,溶解态Fe(Ⅱ)含量在96 h 达到最大值随后迅速降低,而非生物对照组中液相积累的Fe(Ⅱ)没有明显变化。可见体系中微生物作用引起了土壤矿物中的铁氧化物或氢氧化物等发生了还原性溶解,随后被再次固持,在铁的溶解和二次固定过程中土壤固砷矿物基质的形态发生改变,也就是次生矿物对砷的吸附能力降低,这很可能是导致土壤固持的砷发生释放或活化的重要原因。
微生物还原作用下土壤固砷矿物基质的形态发生改变同样造成了土壤固相中铁的活化。与非生物对照相比盐酸可提取的总铁量增加了30.1%。固相中盐酸可提取的Fe(Ⅱ)已达到125.0 mmol·kg-1,约占土壤总铁的20.5%,还原产生的Fe(II)大部分仍固持在固相中。盐酸提取态Fe(T)中盐酸提取态Fe(III)约占38.0%,当微生物还原产生的Fe(II)吸附在未被还原的Fe(III)矿物表面,以减缓Fe(Ⅱ)的释放,此时会形成新生的混合价态铁氧化物矿物,如Fe3O4或者绿锈等混合价态的亚铁矿物[5-6],也可参与固定部分砷。
此外,微生物的硫化作用产物HS-同样会促进铁氧化物/氢氧化物的还原与转化。体系培养168 h后,固相中检测到酸挥发性硫化物的含量已经达到10.3 mmol·kg-1。此时液相Fe(Ⅱ)迅速降低(图4A),盐酸提取态Fe(Ⅱ)含量迅速增加(图4B),可见固相中也存在部分的硫化亚铁,硫离子的存在使液相中溶解的Fe(Ⅱ)的量保持在很低水平。一般认为,新生成的FeS、磁铁矿等次生矿物可以通过吸附、包裹、沉淀或共沉淀等方式固持部分砷,作为盐酸可提取的砷形态中的一个组分。然而本研究采集的土壤培养结果看,土壤矿物的结构转化带来的主要影响仍为砷的活化,与土壤的二次吸附相比次生矿物对砷二次固持的贡献并不大。而且培养后期也逐渐降低,可见这部分矿物固持的As(III)也发生了迁移。由于新生的无定形FeS 将逐渐发生老化并向黄铁矿(FeS2)转化,这一过程,被结合或包裹的砷将变得更加稳定,成为盐酸不可提取的形态。然而目前对于天然复杂体系中无定形硫化亚铁向黄铁矿的转化过程的研究还不多,这一过程仍需进行深入研究。
综上可见,微生物作用下土壤中的铁迅速还原释放后被再次固持,其形态逐渐从结晶的氧化物矿物转化弱结晶或非结晶的形态,亚铁型矿物在土壤中的分配比例逐渐增加,这是导致土壤中砷在不同形态间再分配的重要因素。
3.3 微生物作用下AVS 与砷再分配的关联
厌氧微生物的驱动下,除了发生砷和铁的还原过程,通常伴随着硫的还原过程。硫酸盐还原作用产生的硫化氢与还原产生的二价铁或亚砷酸盐相结合产生铁或砷的硫化物。形成硫化亚铁沉淀时可以同时沉淀或共沉淀体系中释放的砷酸盐和亚砷酸盐。由于硫化亚铁等酸挥发性硫化物对重金属离子的固持具有很重要的作用[22],因此硫酸盐还原过程对砷的环境行为具有强烈影响,并且可在很大程度上干扰控砷的再分配过程。
本研究中厌氧微生物作用下土壤负载的砷发生了还原、释放以及再次固定,通过分离分析溶解态、磷酸盐提取态和盐酸提取态的砷发现,二次固定的砷仅有少部分进入吸附态以及无定形铁氧化物和硫化亚铁结合形态,因此砷的形态可能被转化为其他更稳定的形态。实验在培养过程中检测了固相中酸挥发性硫化物的含量随着培养时间的变化。数据表明,硫离子控制着液相铁和砷的量。体系产生的酸挥发性硫化物的量越多,砷的释放量越少。由于培养的24 h 后释放的As(III)的量已高达15.5 μmol·L-1,此刻极少的溶解态S2-即可形成砷的硫化物沉淀砷。
根据体系中检测的溶解态硫离子和亚砷酸盐浓度计算的离子积KIAP,48 h 后Log KIAP>Log Ksp(As2S3约为10-64.6)[23];48 h 时S(-Ⅱ)和As(Ⅲ)平衡浓度分别为319.0 和304.9 μg·L-1),因此很可能在强烈硫酸盐还原条件下砷以硫化物形态被二次固持,成为盐酸不可提取的形态。在其他学者的研究中也发现在硫酸含量充足的体系中,微生物作用下产生的S(-Ⅱ)会与As(Ⅲ)作用,生成大量的砷硫化物沉淀固定As(Ⅲ)[23-25]。土壤中硫酸盐还原作用可以调控液相中亚铁离子的释放量,并促进砷的固定。但新生的砷的硫化物需要光谱学数据进一步加以证实。
可见土壤微生物作用驱动的铁、砷和硫的还原过程中,土壤中砷的形态转化与二次分配过程是由微生物作用和化学机制共同控制。微生物还原作用造成了土壤所固定的砷的活化或释放,在次生矿物沉淀过程中活化或释放的砷被二次固定。这些过程中土壤负载的砷,其形态从溶解态、吸附态及铁氧化物结合态逐渐被转化为更稳定的硫化物结合态。
贾永锋(1964—),男,博士,研究员,国家杰出青年科学基金获得者,主要研究方向为重金属生物地球化学循环过程与污染控制。
[1] Liu H Y,Probst A,Liao B H.Metal contamination of soils and crops affected by the Chenzhou lead/zinc mine spill(Hunan,China)[J].Science of the Total Environment,2005,339(1-3):153—166
[2] 肖细元,陈同斌,廖晓勇,等.中国主要含砷矿产资源的区域分布与砷污染问题[J].地理研究,2008,27(1):207—208 Xiao X Y,Chen T B,Liao X Y,et al.Regional distribution of arsenic contained minerals and arsenic pollution in China [J].Geographical Research,2008,27(1):207—208(in Chinese)
[3] Vincent C,Remy B,Florence S,et al.Effect of indigenous bacterial activity on arsenic mobilization under anaerobic conditions [J].Environment International,2005,31(2):221—226
[4] Islam F S,Gault A G,Boothman C,et al.Role of metal-reducing bacteria in arsenic release from Bengal delta sediments[J].Nature,2004,430(6995):68—71
[5] Kocar B D,Borch T,Fendorf S.Arsenic repartitioning during biogenic sulfidization and transformation of ferrihydrite[J].Geochimica et Cosmochimica Acta,2010,74(3):980—994
[6] Pedersen H D,Postma D,Jakobsen R.Release of arsenic associated with the reduction and transformation of iron oxides[J].Geochimica et Cosmochimica Acta,2006,70(16):4116—4129
[7] Wang S,Xu L,Zhao Z,et al.Arsenic retention and remobilization in muddy sediments with high iron and sulfur contents from a heavily contaminated estuary in China[J].Chemical Geology,2012,314—317:57—65
[8] Kneebone P E,O'Day P A,Jones N,et al.Deposition and fate of arsenic in iron and arsenic enriched reservior sediments[J].Environmental Science&Technology,2002,36(3):381—386
[9] Lee J,Lee S,Chon H,et al.Enhancement of arsenic mobility by indigenous bacteria from mine tailings as response to organic supply[J].Environment International,2009,35(3):496—501
[10] Samantha L S,Benjamin C B.Changes in iron,sulfur,and arsenic speciation associated with bacterial sulfate reduction in ferrihydrite-rich systems[J].Environmental Science&Technology,2009,43(23):8787—8793
[11] Barringer J L,Mumford A,Young L Y,et al.Pathways for arsenic from sediments to groundwater to steams:Biogeochemical processes in the Inner Coastal Plain,New Jersey,USA[J].Water Research,2010,44(19):5532—5544
[12] Bostick B C,Fendorf S.Arsenic sorption on troilite(FeS)and pyrite(FeS2)[J].Geochimica et Cosmochimica Acta,2003,67(5):909—921
[13] Jonsson J,Sherman D M.Sorption of As(III)and As(V)to siderite,greenrust and magnetite:Implications for arsenic release in anoxic groundwaters[J].Chemical Geology,2008,255(2):173—181
[14] Keon N E,Swartz C H,Brabander D J,et al.Validation of an arsenic sequential extraction method for evaluating mobility in sediments[J].Environmental Science&Technology,2001,35(16):2778—2784
[15] Zhang X,Jia Y,Wang S,et al.Bacterial reduction and release of adsorbed arsenate on Fe(III)-,Al-and coprecipitated Fe(III)/Al-hydroxides[J].Journal of Environmental Sciences,2012,24(3):440—448
[16] 石荣,贾永峰,王承智,等.溶解性有机酸对砷在土壤矿物质表面吸附-解吸行为的影响[J].土壤通报,2009,40(4):761—766 Shi R,Jia Y F,Wang C Z,et al.Effect of dissolved organic acids on arsenate adsorption-desorption onto soil mineral[J].Chinese Journal of Soil Science,2009,40(4):761—766(in Chinese)
[17] Polizzotto M L,Harvey G F,Li G,et al.Solid-phases and desorption processes of arsenic within Bangladesh sediment[J].Chemical Geology,2006,228(13):97—111
[18] Islam F S,Pederick R L,Gault A G,et al.Interactions between the Fe(III)-reducing bacterium Geobacter sulfurreducens and arsenate,and capture of the metalloid by biogenic Fe(II)[J].Applied and Environmental Microbiology,2005,71(12):8642—8648
[19] Pérez-Jiméneza J R,DeFraia C,Young L Y.Arsenate respiratory reductase gene(arrA)for Desulfosporosinus sp.strain Y5[J].Biochemical and Biophysical Research Communications,2005,338(2):825—829
[20] Oremland R S,Stolz J F.The ecology of arsenic[J].Science,2003,300(5621):939—944
[21] Smedley P L,Kinniburgh D G.A review of the source,behaviour and distribution of arsenic in natural waters[J].Applied Geochemistry,2003,17(5):517—568
[22] 北京师范大学、华中师范大学、南京师范大学的无机化学教研室.无机化学上册[M].第四版.北京:高等教育出版社,2002:419—420
[23] Kirk M F,Roden E E,Crossey L J.Experimental analysis of arsenic precipitation during microbial sulfate and iron reduction in model aquifer sediment reactors[J].Geochimica et Cosmochimica Acta, 2010, 74(9):2538—2555
[24] O'Day P A,Vlassopoulos D,Root R,et al.The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions[J].Proceedings of the National Academy of Sciences of the United States of America,2004,101(38):13703—13708
[25] Xu L,Zhao Z,Wang S,et al.Transformation of arsenic in offshore sediment under the impact of anaerobic microbial activities:An in-vitro investigation[J].Water Research,2011,45(20):6781—6788
猜你喜欢
分子催化(2022年1期)2022-11-02 07:11:20
环境保护与循环经济(2021年7期)2021-11-02 08:10:52
食品安全导刊(2021年20期)2021-08-30 06:39:22
皮革制作与环保科技(2020年14期)2020-03-17 07:16:06
当代化工研究(2016年5期)2016-03-20 16:21:35
中国资源综合利用(2016年7期)2016-02-03 03:00:11
环境科技(2015年3期)2015-11-08 12:08:36
电源技术(2015年9期)2015-06-05 09:36:06
中学科技(2015年3期)2015-04-29 04:52:43
河北工业科技(2015年4期)2015-02-27 13:15:36
