
离子色谱法测定食品中的无机铵
2012-12-27 08:50王祖翔
食品与机械 2012年4期
王祖翔 余 杨 孙 莉 蒋 俊
(镇江出入境检验检疫局,江苏 镇江 212008)
离子色谱法测定食品中的无机铵
王祖翔 余 杨 孙 莉 蒋 俊
(镇江出入境检验检疫局,江苏 镇江 212008)
建立一种离子色谱法(IC)快速测定食品中铵根离子含量。样品用水浸泡并混匀,加入乙酸酸化,乙腈沉降蛋白,并一起通过冷冻离心去除脂肪,取适量上清液挥干后用1.0mL水复溶,过膜后备用。以H2SO4溶液梯度淋洗,淋洗液流速为1.0mL/min,采用 DIONEX IonPac CS12A色谱柱分离,带抑制器的电导检测器检测,外标法定量。结果表明:NH+4的线性范围为0.1~20.0mg/L,线性相关系数R2大于0.995,检测限(LOD)为0.05mg/kg,定量限(LOQ)为0.1 mg/kg,加标回收率为74%~110%,其相对标准偏差(RSD)小于5%。该方法简便,灵敏,准确,适用于多种食品中微量铵根离子的检测。
离子色谱;非蛋白氮;铵盐;掺假;定量检测
蛋白质是衡量食品营养价值乃至经济价值的一个重要指标,而中国国家标准规定的蛋白质测定方法仍是基于凯氏定氮法,样品中任何含氮物质均被折算成蛋白质含量,致使食品中非蛋白氮物质掺假得不到有效控制。这不仅危害了人们健康,阻碍了企业生存和行业发展,更影响了中国的国际形象。无机铵盐正是一类非蛋白氮物质,其中含氮量高、性质稳定、价格低廉、应用普遍,且少量加入不影响食品风味的铵盐,极有可能被非法添加到食品中。
目前,常见的铵盐检测方法有:比色法[1]、甲醛法[2,3]、蒸馏法(半微量定氮法)[3,4]等,但是都不同程度的存在难以准确定量、受到游离酸和样品本底色干扰严重和灵敏度低等问题。此外,还有报道采用分光光度法[5]和基于凯氏定氮的新方法研究[6]。离子色谱法[7-10]具有选择性强、灵敏度高、快速简便,稳定性好,可同时测定多组分等优点,但是很少应用于食品中铵离子的检测。
本试验拟建立相关食品中无机铵盐(以NH+4计)定量检测的离子色谱检测法,排除了常见阳离子干扰,为有效监控食品中铵离子是否异常提供了技术支持。
1 材料与方法
1.1 仪器与试剂
硫酸:南京化学试剂有限公司;
Li+(50.0mg/L)、Na+(200.0mg/L)、NH+4(250.0mg/L)、K+(500.0mg/L)、Mg2+(250.0mg/L)、Ca2+(500.0mg/L)混合标准溶液:美国Dionex公司;
NH+4标准溶液:1 000mg/L,美国NSI公司;
一级水:本实验室自制。
离子色谱配有抑制性电导检测器:ICS-3000,美国Di-onex公司;
电子分析天平:AL204,瑞士Mettler Toledo公司;
优普超纯水机:UPH-IV-10T,上海优普纯水仪器设备有限公司;
氮吹仪:TurboVap LV,美国Caliper公司;
涡旋振荡器:XW-80,上海医大仪器厂;
超声波清洗机:SB-1000YDTD,宁波新芝生物科技股份有限公司;
高速冷冻离心机:H2050R,湖南湘仪离心机有限公司。
1.2 色谱条件
色谱柱:DIONEX IonPac CS12A4mm×250mm;保护柱:DIONEX IonPac CG12A4mm×50mm;抑制器:DIONEX CSRS 300 4mm;淋洗液:10mmol/L H2SO4溶液;淋洗程 序:0~18min,2.5mmol/L;18.1~40min,10mmoL/L;40.1~50min;2.5mmol/L;进样量:25μL;流速:1.0mL/min。
1.3 铵离子和干扰离子的混合标准溶液配制
将NH+4(1 000mg/L)标准溶液用一级水稀释为100mg/L和10mg/L储备液,再用一级水稀释得到0.10,0.50,1.00,5.00,10.00,15.00,20.00mg/L的溶液。
将6种阳离子的混标用一级水稀释100倍,得到浓度为Li+(0.50mg/L)、Na+(2.00mg/L)、NH+4(2.50mg/L)、K+(5.00mg/L)、Mg2+(2.50mg/L)、Ca2+(5.00mg/L)的混合标准溶液。
1.4 样品处理
称取试样0.5~1.0g于10mL带塞离心管,加2.0mL 60℃温水,超声提取5min、涡旋,再加入1.0mL乙酸涡旋,确定样品溶液呈酸性,再加入7.0mL乙腈涡旋,-10℃6 000r/min冷冻离心20min,定量吸取上清液4.0mL氮气吹干,加1.0mL一级水溶解,过0.20μm滤膜,备用。
1.5 样品测定
试样经前处理后,按设置的色谱条件测定,采用外标法定量,待测样液中铵根离子的响应值应在标准曲线线性范围内,色谱峰保留时间应与标准物质一致。
2 结果与讨论
2.1 色谱条件的优化
淋洗液的选择与浓度直接影响色谱峰的分离效率和检测的灵敏度。由于铵根离子为阳离子,故选择甲磺酸或硫酸溶液作为淋洗液进行考察。通过试验发现甲磺酸溶液作为淋洗液时,系统的背景电导较高、基线噪音较大,而硫酸溶液作为淋洗液时背景信号低,能获得较高的灵敏度,且淋洗液浓度变化后的平衡时间较短,所以选择H2SO4淋洗液。
试验考察了5种常见的阳离子Li+、Na+、K+、Mg2+、Ca2+对NH+4离子是否存在干扰,图1为10mmol/L H2SO4溶液恒流条件下,6种阳离子混合标准溶液的色谱图。
由图1可知,NH+4离子的色谱峰紧接着Na+离子出峰,出峰时间分别为5.337,4.680min,其分离度小于1.5,不能满足检测实际要求。而Na+离子普遍存在于多种食品中且含量相对较高,对NH+4离子极易造成干扰,通过调整梯度洗脱程序,在目标离子出峰时间段降低淋洗液浓度,增加了NH+4和Na+离子的分离度,使其出峰时间相差达到2.6min,分离度增大到4以上,确保目标离子不被干扰。NH+4离子出峰后,加大淋洗液浓度,缩短检测时间,经过标准品和实际样品试验得到满意结果(见图2)。最终确定了淋洗程序:0~18min,2.5mmol/L;18.1~40min,10mmoL/L;40.1~50min,2.5mmol/L。
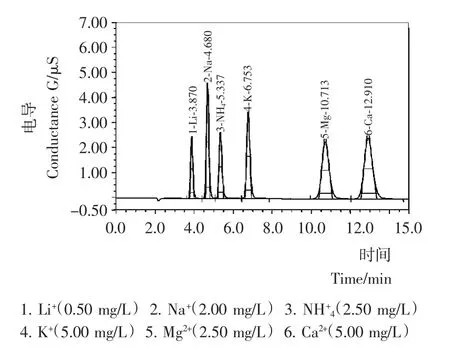
图1 10mmol/L H2SO4浓度下混合标准溶液色谱图Figure 1 Chromatogram of mixed standard solution with 10mmol/L H2SO4
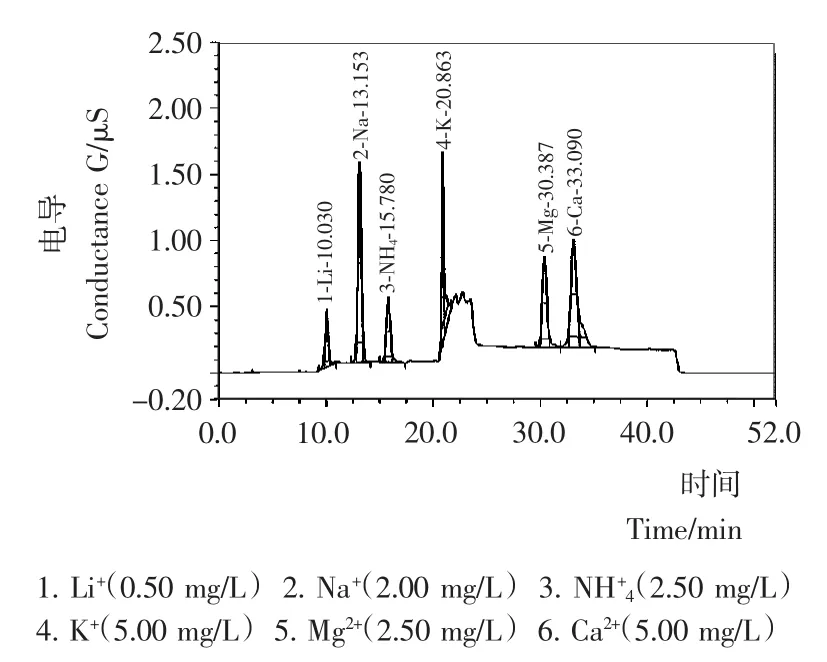
图2 梯度淋洗下混合标准溶液色谱图Figure 2 Chromatogram of mixed standard solution with gradient elution
2.2 线性关系与检出限
以NH+4离子的浓度与其定量离子对应的峰面积绘制标准曲线,采用外标法定量;以10倍的信噪比所对应的待测物质的浓度为最低定量浓度,即S/N=10作为定量限LOQ为0.10mg/L;以3倍信噪比所对应的待测物质的浓度为最低检出浓度,即S/N=3作为检测限LOD为0.05mg/L。NH+4离子在0.1~20.0mg/L浓度范围内的线性方程为Y=0.087 2+0.162 7X、线性相关系数R2=0.998 2。
2.3 样品前处理优化
乙腈作为通用的提取溶剂在残留检测方面得到广泛的应用,且本身对样品中的糖、脂肪和蛋白质的溶解性较小,对蛋白又有沉淀作用,所以试验选用乙腈水溶液作为提取溶剂。通过考察80%,70%,60%乙腈水溶液的沉淀蛋白效果,发现80%乙腈水溶液使蛋白质迅速结块,影响结果的重复性,60%乙腈水溶液沉淀蛋白速度较慢,且冷冻离心去除脂肪效果不佳。
选择效果最好的70%的乙腈水溶液提取,于-10℃冷冻离心去除脂肪后,定量吸取上清液,氮气吹干后用1mL一级水复溶,过0.20μm滤膜供离子色谱检测,其加标回收率几乎为零。通过加入乙酸调节样品处理液呈酸性,再加入乙腈沉淀蛋白后继续进行样品处理,其回收率大幅提高,可满足试验要求。
2.4 实际样品检测结果
选取乳粉和豆浆为实际样品,采用离子色谱技术检测奶粉和豆浆中的铵根离子,按“1.4”的方法进行试样前处理,按“1.2”的色谱条件定量测定铵根离子并进行6次平行试验,获得了良好试验结果,试样检测数据和相对标准偏差见表1,相应试样处理液的色谱图见图3。

表1 实际样品中铵根离子检测数据表Table 1 Detection data and RSDs of NH+4 in actual samples(n=6)
2.5 加标回收率
在10,20,40mg/kg 3个添加水平下,进行准确度试验,计算加标平行样(n=8)的回收率得到:铵根离子添加回收率为73.8%~110.1%,详见表2。
2.6 与标准方法比较
铵离子检测的标准方法有甲醛法和蒸馏法(半微量定氮法)。

图3 实际样品中NH+4色谱图Figure 3 Chromatograms of NH4+in actual samples
甲醛法测定无机铵盐是化学滴定法,在食用氯化钾行业标准中明确了该试验方法。在甲醛法检测NH+4试验中,6mol甲醛与4mol NH+4作用生成1mol质子化的六亚甲基四胺和3mol的H+,可与4mol的OH-直接滴定中和,其反应的摩尔比是1∶1。该方法快速简便,但甲醛毒性高,遇到样品提取液背景色较深时指示剂显色被严重干扰,难以判断反应终点。此外,该方法受到样品自身游离酸干扰非常严重,只适用于基质单一且水溶液为中性的样品检测,根本不能满足食品检验需求,故没有对该方法进行试验考察。
蒸馏法测定无机铵盐是半微量定氮法,在酱油卫生标准中明确了该试验方法,本课题以奶粉为试样,只对蒸馏法测定铵盐进行了考察。
称取试样0.5~1.0g于1 000mL圆底烧瓶,加300mL水溶解,连接蒸馏装置后检查气密性,再加入45%氢氧化钠溶液5mL加热进行试验。使用20mL 2%硼酸溶液作为吸收液,0.05mol/L盐酸标准滴定溶液作为标准滴定溶液。
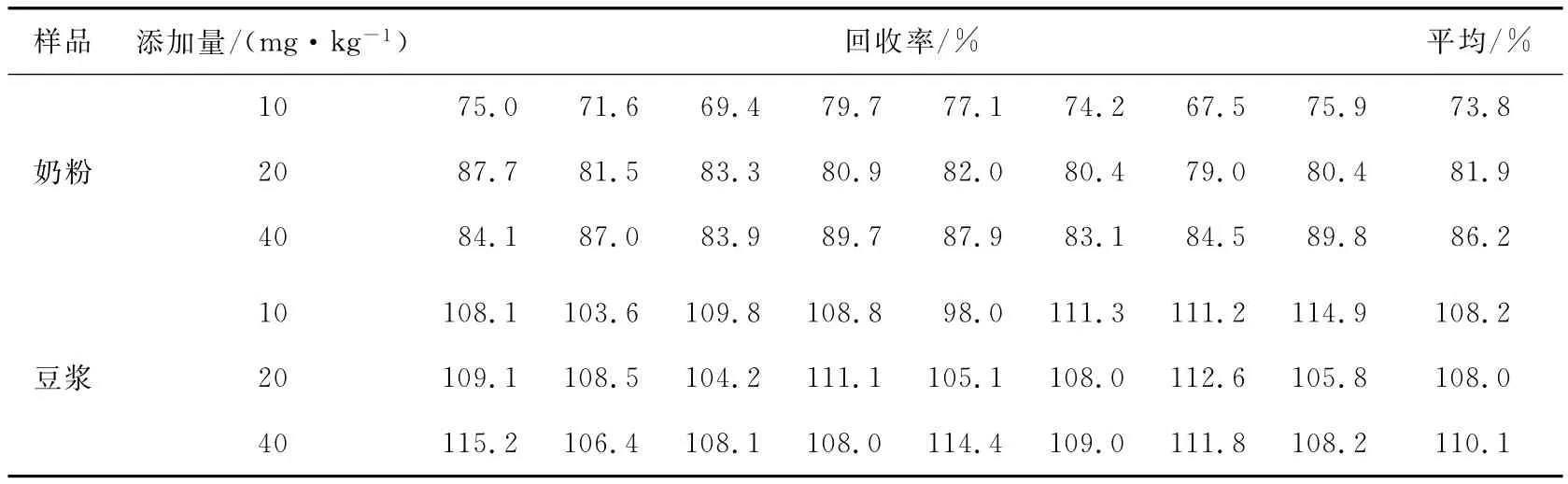
表2 铵离子回收率Table 2 Recoveries of NH+4spiked at different levels(n=8)
试验得到20个检测数据,计算得到精密度为9%。以空白的标准偏差为信号噪音,3倍的标准偏差为最低检出限,通过计算为20mg/kg。试样中定量添加硫酸铵或氯化铵水溶液,进行加标回收试验,计算加标回收率为93%~103%。
试验结果表明:该方法稳定性和准确度良好,但是灵敏度很低,检测周期较长,操作繁琐。由于样品不经过消解,大量的有机物会在蒸馏装置中发生严重的鼓泡现象,直接影响检测结果,无法满足当今食品非法添加物残留检测的要求。
3 结论
本试验采用离子色谱技术,建立了食品中铵根离子的检测方法,把硫酸铵、氯化铵和硝酸铵等一系列无机铵盐打包处理,有效的监控食品中铵根离子浓度是否异常,从而能够判断食品中是否存在人为掺假无机铵盐的风险。
1 弓晓峰,张静,张振辉,等.纳氏试剂比色法测定土壤铵态氮的研究[J].环境科学与技术,2006,29(1):43~44.
2 中华人民共和国国家经济贸易委员会.QB 2554——2002食用氯化钾[S].北京:中国轻工业出版社,2002.
3 陆思伟,吕翔,王岩,等.蒸馏法和甲醛法测定高氯酸铵含量的比较[J].无机盐工业,2010,42(8):60~61.
4 中华人民共和国卫生部,中国国家标准化管理委员会.GB/T 5009.39——2003酱油卫生标准的分析方法[S].北京:中国标准出版社,2004.
5 Jota Kanda.Determination of ammonium in seawater based on the indophenol reaction with o-phenylphenol(OPP)[J].Water Research,1995,29(12):2 746~2 750.
6 Xiao-Li Su,Li-Hua Nie,Shou-Zhuo Yao.Determination of ammonium in Kjeldahl digests by gas-diffusion flow-injection analysis with a bulk acoustic wave-impedance sensor[J].Talanta,1997,44(11):2 121~2 128.
7 钟志雄,李攻科,朱炳辉,等.离子色谱法同时测定化妆品中的铵和6种烷基胺[J].色谱,2010,28(7):702~707.
8 关良智,于泓.单柱离子色谱法同时测定碱金属、铵离子和胺类[J].色谱,1995,13(1):53~54.
9 Christopher Pohl,Maria Rey,Detlef Jensen,et al.Determination of sodium and ammonium ions in disproportionate concentration ratios by ion chromatography[J].Journal of Chromatography A,1999,850(1~2):239~245.
10 Frédéric Gaucheron,Yvon Le Graet.Determination of ammonium in milk and dairy products by ion chromatography[J].Journal of Chromatography A,2000,893(1):133~142.
Studies on determination of inorganic ammonia in foods by ion chromatography
WANG Zu-xiang YU Yang SUN LiJIANG Jun
(ZhenJiang Entry-Exit Inspection and Quarantine Bureau,Zhenjiang,Jiangsu212008,China)
A new method of ion chromatography(IC)was developed for the determination of inorganic ammonia in foods.Samples were soaked and mixed in water,acidified by acetic acid,and proteins were precipitated by adding acetonitrile and removed with fat by freezing centrifugation.Then the quantitative supernatant was evaporated to dry and the residues were filtrated with membrane and reserved for analysis after dissolved in 1mL water.The H2SO4solution was selected as the gradient elution at a flow rate of 1.0mL/min.The sample solution was separated by column DIONEX IonPac CS12A,and then detected by conductivity detector with suppressor.And external standard curve was used for the need of quantitative analysis.Results:The linear range of the method for NH+4was 0.1~20.0mg/L,and the linear correlation coefficientR2was more than 0.995.The limit of detection(LOD)was 0.05mg/kg and the limit of quantitation(LOQ)was 0.1mg/kg.The recoveries were 74%~110%with the relative standard deviations(RSDs)less than 5%.Actual test results showed that the method was simple,sensitive and accurate,and was suitable for trace determination of NH+4in many foods.
ion chromatography(IC);non-protein nitrogen;ammonia;adulteration;quantitative detection
10.3969 /j.issn.1003-5788.2012.04.026
江苏出入境检验检疫局项目(编号:2009KJ35)
王祖翔(1983-),男,镇江出入境检验检疫局技术中心工程师。E-mail:wang_zuxiang@163.com
2012-01-15
猜你喜欢
煤化工(2022年3期)2022-07-08
世界科学技术-中医药现代化(2020年2期)2020-07-25
中成药(2018年12期)2018-12-29
中成药(2017年6期)2017-06-13
中国资源综合利用(2016年10期)2016-01-22
广州大学学报(自然科学版)(2015年4期)2015-12-23
医学研究杂志(2015年4期)2015-06-10
丝绸(2015年11期)2015-02-28
中国洗涤用品工业(2012年4期)2012-03-20
中国洗涤用品工业(2011年4期)2011-03-20
