甲醇对1-丁基-3-甲基咪唑四氟硼酸离子液体结构与性质影响的模拟研究
2012-12-11 09:06田国才
物理化学学报 2012年11期
王 丁 田国才
(昆明理工大学冶金与能源工程学院,昆明650093)
甲醇对1-丁基-3-甲基咪唑四氟硼酸离子液体结构与性质影响的模拟研究
王 丁 田国才*
(昆明理工大学冶金与能源工程学院,昆明650093)
采用分子动力学模拟方法研究了298.15 K、0.1 MPa下摩尔分数为0.1-0.9的甲醇对1-丁基-3-甲基咪唑四氟硼酸盐([BMIM][BF4])结构与性质的影响.获得了体系的密度、径向分布函数、配位数、自扩散系数、粘度和电导率,模拟得到的密度值与实验值符合较好.结果显示:体系各组分之间的径向分布函数随甲醇摩尔分数的增加呈规律性变化;体系内阴阳离子的自扩散系数随着甲醇摩尔分数的增加不断增大;甲醇的加入削弱了阴阳离子之间的相互作用,体系粘度随着甲醇摩尔分数的增加逐渐减小,电导率不断增大.分析空间分布函数得到体系中各组分的三维空间分布情况.
离子液体;1-丁基-3-甲基咪唑四氟硼酸盐;甲醇;分子动力学模拟;微观结构;物理化学性质
1 引言
离子液体,又称室温离子液体或室温熔盐,是完全由特定的阳离子和阴离子构成的在室温或近于室温下呈液态的离子体系.它具有电化学窗口宽、导热导电良好、不易挥发、不可燃、热稳定性好等一系列特点而被誉为新型“绿色”溶剂和电解质,已经广泛和成功地应用于材料制备、催化、金属电沉积、萃取分离、有机合成、燃料电池等领域.1-5然而,研究发现离子液体中其它物质的存在对离子液体的结构、物理和化学性质有极大影响.6事实上,离子液体的合成过程,不仅要使用大量的有机试剂、溶剂和水,而且合成过程中的副反应也会产生其他的有机物质,同时离子液体的提纯不可避免地要使用有机溶剂.研究表明这些物质的存在会极大地影响离子液体的结构、物理和化学性质,同时还会影响离子液体自身的溶剂性质和输运性质,进而影响发生在离子液体内反应的速率和选择性.6因此,研究常用有机分子对离子液体结构与性质的影响对于离子液体的设计、合成以及工业应用至关重要.
近年来,离子液体与有机分子组成多元体系的研究已见诸报道.实验方面,中国科学院化学研究所Han等7测定了288.15-308.15 K时溶质(水+乙醇/丙酮)摩尔分数为0.2-0.48的1-丁基-3-甲基咪唑六氟磷酸盐([BMIM][PF6])/水/乙醇以及[BMIM][PF6]/水/丙酮三元复合体系的电导率和粘度.结果表明,随着溶质摩尔分数的增加,体系的电导率不断增大,温度升高和溶质摩尔分数增加则体系的粘度减小.河南师范大学王键吉等8运用核磁共振研究了1-辛基-3-甲基咪唑四氟硼酸盐([OMIM][BF4])离子液体与丙酮分子之间的相互作用,结果表明阳离子芳环上的氢与氮直接相连的甲基和亚甲基上的氢与丙酮羰基上的氧具有较强的相互作用,从而减弱了离子液体[OMIM][BF4]阴阳离子之间的相互作用,使离子对的运动加快,粘度降低.Arce等9测定了[OMIM][BF4]分别与甲醇、乙醇、正丙醇和异丙醇组成的二元体系在298.15 K时的密度、折射率、声速和动态粘度,在此基础上计算得到的过量摩尔体积、摩尔折射率和等熵压缩性符合Redlich-Kister多项式.Zafarani-Moattar和Majdan-Cegincara10报道了298.15 K时[BMIM][PF6]分别与甲醇和乙腈组成体系的粘度、折射率以及密度.Domanska等11测定了298.15-348.15 K下1-丁基-3-甲基咪唑硫氰酸盐([BMIM][SCN])分别与甲醇、乙醇、丙醇组成的二元体系的密度与粘度,结果表明醇中碳链的长度对体系的物理性质有很大影响.侯海云等12报道了293.15 K下1,3-二甲基咪唑醋酸盐([DMIM][CH3COO])、1-乙基-3-甲基咪唑醋酸盐([EMIM][CH3COO])分别与水和乙醇组成的二元体系在全浓度范围内的电导率,计算了相应咪唑醋酸盐的摩尔电导率,发现无论是水溶液还是乙醇溶液,溶液的电导率和咪唑醋酸盐的摩尔电导率都随水和乙醇浓度的增加先增大后减小.
纵观近年实验进展可以发现,大多数文献集中于离子液体与有机溶剂组成的多元体系的热力学性质以及输运性质的测定,有关离子液体与有机分子体系各组分之间的相互作用以及有机分子的存在如何影响离子液体的微观结构和相关性质的实验研究工作还很少.近年来,分子动力学模拟技术迅速崛起,它可以从微观上系统地研究物质的结构、热力学性质以及原子扩散等动力学性质,可获得大量实验难以得到的重要信息.Hanke等13对氯化1,3-二甲基咪唑([DMIM][Cl])分别与水、甲醇、二甲醚和丙烷组成的二元体系进行了分子动力学模拟研究,结果发现氯阴离子主要与水和甲醇形成氢键,而在与二甲醚以及丙烷的混合体系中氯阴离子主要与阳离子发生相互作用.北京化工大学刘志平14和Wang课题组15在其前期开发的力场之上对[BMIM][BF4]与水、乙腈体系进行了模拟,分析了体系组分之间的径向分布函数和空间分布情况,得到的密度、过量摩尔体积与实验值符合得很好,表明该力场能很好地描述混合体系内各组分之间的相互作用.Raabe和Kohler16研究了氯化1-烷基-3-甲基咪唑([AMIM][Cl])系列离子液体分别与乙醇、丙醇组成的二元混合体系,得到了混合体系的密度、过量混合能、径向分布函数以及空间分布函数,发现氯阴离子与醇中羟基之间的氢键相互作用随离子液体中阳离子烷基碳链增长而增大.Jahangiri等17采用分子动力学模拟方法研究了氯化1-乙基-3-甲基咪唑([EMIM][Cl])、1-乙基-3-甲基咪唑六氟磷酸盐([EMIM][PF6])分别与甲醇和乙醇组成的二元混合体系的过量性质以及结构性质.结果表明甲醇倾向于与离子液体中的阴离子部分聚合.Mendez-Morales等18报道了1-己基-3-甲基咪唑四氟硼酸盐([HMIM][BF4])、1-己基-3-甲基咪唑六氟磷酸盐([HMIM][PF6])和氯化1-己基-3-甲基咪唑([HMIM] [Cl])分别与甲醇和乙醇组成的二元混合体系的密度、过量摩尔体积、整体和局部的径向分布函数、配位数和氢键的模拟工作.研究发现阴离子的性质和醇分子体积对聚合过程有极大影响,比较阴离子与甲醇、阴离子与乙醇、阳离子与甲醇以及阳离子与乙醇之间的径向分布函数的第一峰值发现醇类分子优先与离子液体中阴离子发生相互作用,特别是甲醇与阴离子为卤素离子的离子液体混合体系中.
作为咪唑类离子液体重要一员的[BMIM][BF4]凭借其一系列优点广泛应用于有机合成、材料制备、催化和电化学等领域.6在[BMIM][BF4]离子液体的合成过程中常常要用到甲醇等有机溶剂,19研究表明[BMIM][BF4]离子液体能与甲醇互溶,20而有机分子的存在如何影响离子液体微观结构及物理化学性质尚不清楚.因此考察甲醇等有机分子对离子液体[BMIM][BF4]微观结构以及性质的影响,可为[BMIM][BF4]的大规模合成以及工业应用、运输和储存提供理论指导和依据.本文通过分子动力学模拟研究了298.15 K、0.1 MPa下,摩尔分数为0.1-0.9的甲醇对1-丁基-3-甲基咪唑四氟硼酸盐([BMIM] [BF4])结构与性质的影响,得到了体系密度值,对比实验值验证了所用力场的准确性;考察体系的径向分布函数、配位数以及空间分布函数;分析了甲醇的加入对体系粘度、电导率以及扩散系数的影响,进一步揭示了体系各组分之间相互作用的微观本质.
2 力场
本文涉及的离子液体[BMIM][BF4]及甲醇的结构由量子化学中密度泛函理论方法B3LYP/6-311++ (d,p)计算得到,如图1所示.图中各原子的符号与其在力场(参见Supporting Information)中的标识完全一致.模拟采用Canongia-Lopes等21,22开发的全原子力场,该力场已成功应用于不同体系的分子动力学模拟研究,23-27其函数形式如下:


图1 离子液体[BMIM][BF4]与甲醇的结构Fig.1 Molecular structures of ionic liquid[BMIM][BF4] and methanol
式(1-6)中,Utotal表示体系的总能量;Ustretch和Ubend分别为键伸缩项和键角弯曲项,二者均以简谐势表示,力常数分别为Kr和Kθ,平衡键长及键角为r0和θ0;二面角扭矩能Utorsion以余弦函数形式表示,Vi为傅里叶系数,ϕ为二面角;ULJ和Ucoulomb分别表示范德华作用能和静电相互作用能,qi和qj为原子i与j所带的电荷数,rij为原子i和原子j之间的距离.原子间的Lennard-Jones参数σij和εij由Lorentz-Berthelot组合规则得到.与大多数离子液体体系模拟的文献23-27一致,本文阳离子[BMIM]+参数取自Canongia-Lopes等21,22基于OPLS-AA和AMBER力场针对烷基咪唑阳离子开发的全原子力场,该力场烷基碳链长度可延展至十多个碳原子.阴离子参数取自于de Andrade等28开发的力场,甲醇分子参数取自Cornell等29开发的力场,其二面角扭曲项形式为:

式中Vn为傅里叶系数,γ为扭转角.鉴于篇幅,相关力场参数在Supporting Information中给出.
3 模拟体系和细节
分子动力学模拟采用MDynaMix程序,30模拟体系及粒子数详见表1,甲醇摩尔分数x1分别为0.1, 0.2,0.3,0.4,0.5,0.6,0.7和0.9.将阴阳离子对和甲醇分子随机放入模拟盒子中作为体系的初始构型,采用周期性边界条件,截断半径均为1.5 nm(纯甲醇体系为1.3 nm),静电相互作用采用Ewald求和技术处理.运动方程采用Tuckerman-Berne双时间步长算法求解,长时间和短时间步长分别为2和0.2 fs.体系温度和压力分别保持为298.15 K和0.1 MPa不变,耦合常数分别为30和700 fs,首先在NPT系综下运行400 ps至总能量收敛,密度达到平衡,然后以NPT系综模拟得到的最终平衡构型为NVT系综的初始构型,再运行2 ns计算所需要的物理量,每5步储存构象文件用于后续分析,NVT系综模拟的盒子大小在表1中给出.

表1 模拟体系的大小及T=298.15 K,p=0.1 MPa下计算得到的密度Table 1 System sizes of different simulation runs and simulated densities at T=298.15 K,p=0.1 MPa
4 结果与讨论
4.1 密 度
密度是验证力场是否准确可靠的重要依据.本文模拟得到的纯离子液体[BMIM][BF4]与甲醇在298.15 K、0.1 MPa下的密度(见表1)分别为1.169和0.7543 g·cm-3,与该温度下的实验值(1.201和0.7865 g·cm-3)31符合较好.图2中给出了模拟得到的[BMIM][BF4]/CH3OH体系的密度与实验值31的对比.从图中可以看出,模拟得到[BMIM][BF4]/CH3OH体系的密度与实验值符合较好.这进一步表明,本文所用的力场能够较好地描述体系中各组分间的相互作用,可以较准确地预测体系的性质.
4.2 径向分布函数和配位数
径向分布函数(RDF)是表征流体局部微观结构的重要函数,14可由下式求得,

其中N为体系总原子数,ρ=N/V,为体系的平均数密度,<…>表示系综平均.
图3(a-d)分别给出了阳离子五元环中CR与阴离子中B原子、阳离子五元环中CR与甲醇羟基O原子、阴离子中F原子与甲醇羟基O原子以及甲醇羟基O原子之间的径向分布函数,其中图3(c)中小图为阴离子中F原子与甲醇羟基O原子之间的径向分布函数第一峰的放大图.为了计算和讨论上的方便,我们分别用CR代表阳离子,B代表阴离子,O代表甲醇分子.为了便于分析粒子之间的相互作用,表2给出了模拟得到的不同体系中各粒子之间的相互作用能.

图2 模拟得到的密度与实验值31的对比Fig.2 Comparison between simulation and experimental density31values
从图3(a)可以看出随着甲醇摩尔分数的增加阳离子五元环中CR原子与阴离子中B原子之间的径向分布函数第一峰出峰位置不断右移,结合表2给出的阴阳离子之间的相互作用能可知阴阳离子之间的相互作用随着甲醇摩尔分数的增加不断降低,不断增加的甲醇分子占据了[BMIM]+周围BF-4的位置,导致第一峰的出峰位置随着甲醇摩尔分数的增加不断增大.分析图3(b)中CR与O之间的RDF发现在r=0.339,0.543 nm处分别出现第一峰和第二峰,RDF最高峰的峰值小于2且在r约为0.6 nm之后RDF趋于平缓.综合上述情况得出阳离子周围并未出现甲醇分子的聚合层,类似的情况也出现在Jahangiri等17模拟报道的[EMIM][PF6]、[EMIM][Cl]分别与甲醇组成的混合体系以及Mendez-Morales等18研究的[HMIM][PF6]、[HMIM][BF4]、[HMIM][Cl]分别和甲醇形成的混合物中.为更好地反映阴离子与甲醇分子的相互作用关系,图3(c)选取BF-4中的氟原子代表阴离子.阴离子与甲醇分子间的径向分布函数中第一峰峰值随着甲醇摩尔分数增加顺序增大,出峰位置为r=0.267 nm,表明阴离子与甲醇分子之间形成了氢键,并且随着甲醇摩尔分数的增加阴离子与甲醇之间的氢键相互作用逐渐增强.图3(d)表示的是甲醇羟基O原子之间的径向分布函数,从图中第一峰的出峰位置r=0.273 nm得出甲醇分子之间存在氢键相互作用,并随着甲醇摩尔分数增加氢键相互作用增强.此外,r=0.465 nm时出现第二峰表明在甲醇分子周围形成了两个距离较近的甲醇分子聚合层.
从以上分析得知甲醇的加入削弱了阴阳离子相互作用,甲醇与阴离子之间形成氢键,同时与阳离子发生离子-偶极相互作用.上述结论也可以通过对配位数的分析得到.配位数是指距离中心原子一定距离范围内目标原子的数目.若取径向分布函数图的第一波谷出现的距离作为第一溶剂层的厚度,则该溶剂层内目标原子的配位数可由下式求出,32
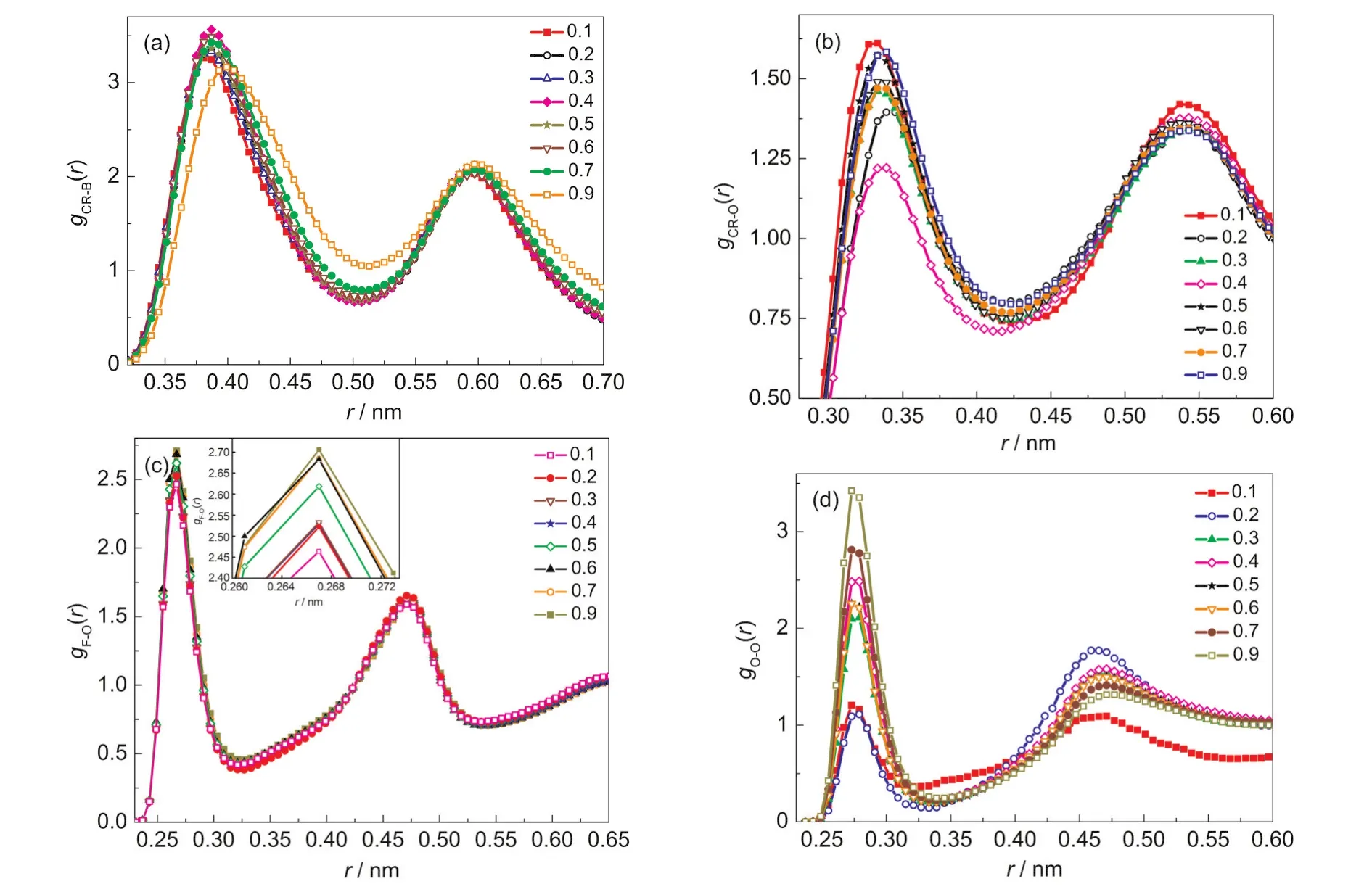
图3 体系中不同原子间的径向分布函数Fig.3 Radial distribution functions between different atoms(a)CR(cation ion)in the imidazolium ring and B atom in ,(b)CR in the imidazolium ring and O atom in methanol, (c)F atom inand O atom in methanol,(d)O atom in different methanols.For the notations,see Fig.1.

其中ρ0表示目标原子的平均密度,g(r)为径向分布函数值,r为第一波谷的出现位置.
图4给出了阳离子周围的阴离子与甲醇、阴离子周围的甲醇以及甲醇分子之间的配位数与甲醇摩尔分数的关系.从图中可以看出随着甲醇摩尔分数的增加,阳离子周围阴离子的数目不断减少,同时阳离子和阴离子周围的甲醇配位数以及甲醇分子周围的甲醇配位数不断增加.结合表2,不难看出这是因为随着甲醇摩尔分数的增加阴阳离子之间的相互作用逐渐减弱,阳离子和阴离子与甲醇的相互作用以及甲醇分子之间的相互作用不断增强,这与从径向分布函数的角度分析得到的结果是一致的.比较阳离子与阴离子周围甲醇配位数以及表2中阴阳离子分别与甲醇分子之间的相互作用能可发现,甲醇摩尔分数相同时,阳离子周围甲醇的配位数要少于阴离子,同时阴离子与甲醇之间相互作用要强于阳离子与甲醇分子之间的相互作用,说明了甲醇的加入在削弱阴阳离子相互作用的同时,与阴阳离子发生了相互作用,且与阴离子的相互作用要强于阳离子.

表2 分子动力学模拟得到的各粒子之间的相互作用能Table 2 Intermolecular energies of different components from molecular dynamics simulations

图4 T=298.15 K,p=0.1 MPa下阳离子与周围的阴离子、甲醇和阴离子与周围的甲醇以及甲醇周围其它甲醇的配位数Fig.4 Coordination numbers of cation-anion,cationmethanol,anion-methanol,methanol-methanol at T=298.15 K,p=0.1 MPa
4.3 自扩散系数
离子液体的自扩散系数(D)33是表征离子液体扩散动力学特性的一个重要参数,可由Einstein方程33计算得到

式中[ri(t)-ri(0)]2为粒子i的均方根位移,〈…〉为系综平均,ri(t)表示时间t时粒子i的位置.
图5给出了阳离子[BMIM]+、阴离子以及甲醇的自扩散系数随甲醇摩尔分数的变化关系.从图中可明显看出随着甲醇摩尔分数的增加,阴阳离子以及甲醇的自扩散系数不断增加,但并非线性关系.在甲醇摩尔分数较低时,阴阳离子以及甲醇移动较为迟缓,这是因为在三种粒子之间的相互作用比较强,而在x1>0.6时,三者的自扩散系数迅速增大,说明了甲醇的大量加入迅速削弱了阴阳离子之间的相互作用导致体系各组分扩散加快,粘度下降,电导率增大.此外,阴阳离子的自扩散系数值十分接近,但阳离子的自扩散系数值始终大于阴离子.尽管阴离子的尺寸更小,但由于阴离子与甲醇的相互作用要大于与阳离子与甲醇之间的相互作用,从而导致了阳离子的自扩散系数大于阴离子.33

图5 T=298.15 K,p=0.1 MPa不同甲醇摩尔分数下阳离子[BMIM]+、阴离子以及甲醇的自扩散系数(D)Fig.5 Self-diffusion coefficient(D)of the[BMIM]+cation, anion,and methanol at different methanol mole fractions at T=298.15 K,p=0.1 MPa
4.4 粘度与电导率
粘度作为离子液体的重要物理参数,其大小对其传质方式、动力的选择具有很大影响.粘度通常可以根据Stokes-Einstein关系式34计算得到,

式中D+为阳离子扩散系数,kB为玻尔兹曼常数(取1.38×10-23J·K-1),R+为阳离子[BMIM]+水力学半径(3.55×10-10m).35电导率是离子液体能否在电化学中应用的重要指标,同时还可以用来解释电解质的电化学行为,可根据Nernst-Einstein方程36求得,

式中N为单位体积阴阳离子数目,q为电子电荷,D-为阴离子扩散系数.

图6 甲醇摩尔分数对体系粘度(η)与电导率(κ)的影响Fig.6 Effect of methanol mole fraction on viscosities(η) and conductivities(κ)of the system
图6给出了甲醇摩尔分数对体系粘度与电导率的影响,通过式(11)和(12)计算得到的粘度与电导率值与文献37报道的实验值符合较好.从图6(a)中可以看出随着甲醇摩尔分数的增加体系的粘度不断减小,结合上文中RDF分析可知加入的甲醇与阴离子形成了氢键同时与阳离子发生离子-偶极相互作用,从而削弱了阴阳离子之间的相互作用,体系中离子淌度增大,体系的粘度随之减小.图6(b)为体系的电导率随甲醇摩尔分数变化关系图.一般来说,离子液体的电导率与粘度呈反比,粘度越大,导电性能越差.从图6可以看出模拟体系的粘度和电导率呈明显的反比关系,甲醇的加入与离子液体的阴离子形成氢键,同时与阳离子发生离子-偶极相互作用.这些相互作用削弱了离子液体阴阳离子的缔合,增加了溶液中带电粒子数,提高了离子的淌度和离子电导率.38
4.5 空间分布函数
空间分布函数直观地描述了体系中各组分在三维空间的分布情况.使用gOpenMol软件39通过等密度面来实现中心分子周围任意分子的三维分布.图7和图8为x1=0.2时体系中各物种的空间分布函数.图7(a)表示的是阳离子[BMIM]+周围阴离子BF-4的三维空间分布情况,从图中可以看出阴离子主要聚集在CR(参看图1中原子的定义,下同)周围,这主要是因为CR正电荷的密度较大.其次在CW与烷基链之间的区域也有阴离子的分布,同时由于丁基基团的排斥作用,阴离子密度为平均密度15倍的区域分布在CW与甲基之间.此外,在靠近丁基尾端可以发现阴离子的聚集,此为对应阴阳离子之间RDF图中阴离子在阳离子周围的第二聚合壳层.图7(b)所示的甲醇分子在阳离子周围空间的分布情况类似于阴离子在阳离子[BMIM]+周围空间的分布,均集中在CR周围以及CW与烷基链之间的区域,但在甲醇分子区域密度为平均密度的3倍时,甲醇分子并未在阳离子[BMIM]+周围形成紧密的聚合壳层,这与从RDF角度分析得到的结果是一致的.观察图8(a)甲醇分子在阴离子周围的分布,可以看出甲醇分子均匀地分布在阴离子中四个F原子组成的四面体的四个顶点之上.另外,从图8(b)甲醇分子周围其它甲醇的空间分布图可以得知在甲醇区域密度为平均密度的8倍时,甲醇分子主要分布在羟基周围,这是因为甲醇分子之间形成了强烈的氢键相互作用,致使甲醇分子在此区域的密度增大.区域密度为平均密度的3倍时,在甲醇甲基周围存在大面积的其他甲醇分子,此为对应的甲醇分子之间RDF图中甲醇分子的第二聚合层.
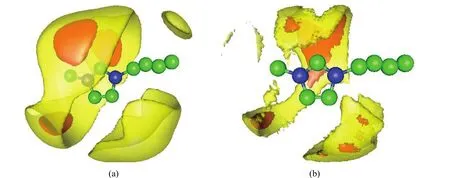
图7 阳离子[BMIM]+周围的(a)阴离子以及(b)甲醇分子三维空间分布Fig.7 Three-dimensional probability distributions of(a)around[BMIM]+and(b)CH3OH around[BMIM]+(a)The orange and yellow bounded contour surfaces are drawn at 15 and 3 times the average density,respectively;(b)the orange and yellow bounded contour surfaces are drawn at 5 and 3 times the average density,respectively.

图8 甲醇在(a)阴离子以及(b)甲醇周围的三维空间分布Fig.8 Three-dimensional probability distribution of(a)CH3OH around and(b)CH3OH around CH3OH(a)The orange and yellow bounded contour surfaces are drawn at 5 and 3 times the average density,respectively;(b)the orange and yellow bounded contour surfaces are drawn at 8 and 3 times the average density,respectively.
5 结论
采用分子动力学模拟方法研究了摩尔分数为0.1-0.9的甲醇对[BMIM][BF4]离子液体结构与性质的影响.模拟所得的体系密度随着甲醇摩尔分数的增加不断减小,与实验值符合较好.分析体系中各组分之间的径向分布函数及各物种之间的相互作用能显示,随着甲醇摩尔分数的增大,阴阳离子之间的相互作用减弱,阴离子与甲醇分子、阳离子与甲醇分子以及甲醇分子之间的相互作用不断增强.随着甲醇摩尔分数的增加,体系中阴阳离子的自扩散系数不断增大,粘度不断减小,电导率不断增大. RDF分析表明阴离子以及甲醇在阳离子周围的分布情况类似,但由于丁基的排斥作用,阴离子主要分布于阳离子甲基一侧,甲醇均匀分布于阴离子四面体的四个顶点上,甲醇羟基附近粒子密度远高于平均密度表明甲醇分子之间形成了强烈的氢键相互作用.
Supporting Information: The force fields parameters for [BMIM]+,BF-4,and methanol have been included.This information is available free of charge via the internet at http://www. whxb.pku.edu.cn.
(1) Dupont,J.;de Souza,R.F.;Suarez,P.A.Z.Chem.Rev.2002, 102,3667.doi:10.1021/cr010338r
(2) Earle,M.J.;Seddon,K.R.Pure Appl.Chem.2000,72,1391. doi:10.1351/pac200072071391
(3) Wilkes,J.Green Chem.2002,4,73.doi:10.1039/b110838g
(4) Hagiwara,R.;Ito,Y.J.Fluor.Chem.2000,105,221.doi: 10.1016/S0022-1139(99)00267-5
(5) Wakai,C.;Oleinikova,A.;Ott,M.;Weingartner,H.J.Phys. Chem.B 2005,109,17028.doi:10.1021/jp053946+
(6)Welton,T.Chem.Rev.1999,99,2071.doi:10.1021/cr980032t
(7)Zhang,J.M.;Wu,W.Z.;Jiang,T.;Gao,H.X.;Liu,Z.M.;He, J.;Han,B.X.J.Chem.Eng.Data 2003,48,1315.doi:10.1021/ je034078h
(8) Zhai,C.P.;Wang,J.J.;Xuan,X.P.;Wang,H.Q.Acta Phys.-Chim.Sin.2006,22,456. [翟翠萍,王键吉,轩小朋,汪汉卿.物理化学学报,2006,22,456.]doi:10.3866/PKU. WHXB20060413
(9) Arce,A.;Rodil,E.;Soto,A.J.Solut.Chem.2006,35,63. doi:10.1007/s10953-006-8939-y
(10) Zafarani-Moattar,M.T.;Majdan-Cegincara,R.J.Chem.Eng. Data 2007,52,2359.doi:10.1021/je700338t
(11)Domanska,U.;Laskowska,M.J.Chem.Eng.Data 2009,54, 2113.doi:10.1021/je8008254
(12)Hou,H.Y.;Huang,Y.R.;Wang,S.Z.;Bai,B.F.Acta Phys.-Chim.Sin.2011,27,2512. [侯海云,黄银蓉,王升泽,白博峰.物理化学学报,2011,27,2512.]doi:10.3866/PKU. WHXB20111120
(13)Hanke,C.G.;Atamas,N.A.;Lynden-Bell,R.M.Green Chem. 2002,4,107.doi:10.1039/b109179b
(14) Wu,X.P.;Liu,Z.P.Acta Phys.-Chim.Sin.2005,21,1036. [吴晓萍,刘志平.物理化学学报,2005,21,1036.]doi:10.3866/ PKU.WHXB20050918
(15)Wu,X.P.;Liu,Z.P.;Huang,S.P.;Wang,W.C.Phys.Chem. Chem.Phys.2005,7,2771.
(16) Raabe,G.;Kohler,J.J.Chem.Phys.2008,129,144503.doi: 10.1063/1.2990653
(17) Jahangiri,S.;Taghikhani,M.;Behnejad,H.;Ahmadi,S.J.Mol. Phys.2008,8,1015.doi:10.1080/00268970802068495
(18) Mendez-Morales,T.;Carrete,J.;Cabeza,O.;Gallego,L.J.; Varela,L.M.J.Phys.Chem.B 2011,115,11170.doi:10.1021/ jp206341z
(19) Ye,T.X.;Zhang,Y.H.;Liu,J.H.;Zhang,Z.L.J.China Univ. Petro.(Edition of Natural Science)2004,28(4),105. [叶天旭,张予辉,刘金河,张在龙.中国石油大学学报(自然科学版),2004,28(4),105.]
(20) Zhao,D.B.;Kou,Y.Univ.Chem.2002,17(1),42.[赵东滨,寇 元.大学化学,2002,17(1),42.]
(21) Canongia-Lopes,J.N.;Deschamps,J.;Padua,A.A.H.J.Phys. Chem.B 2004,108,2038.doi:10.1021/jp0362133
(22) Canongia-Lopes,J.N.;Deschamps,J.;Padua,A.A.H.J.Phys. Chem.B 2004,108,11250.doi:10.1021/jp0476996
(23) Bhargava,B.L.;Balasubramanian,S.J.Chem.Phys.2005,123, 144505.doi:10.1063/1.2041487
(24) Schroder,C.;Rudas,T.;Neumayr,G.;Benkner,S.;Steinhauser, O.J.Chem.Phys.2007,127,234503.doi:10.1063/1.2805074
(25) Kowsari,M.H.;Alavi,S.;Ashrafizaadeh,M.;Najafi,B. J.Chem.Phys.2008,129,224508.doi:10.1063/1.3035978
(26) Skarmoutsos,I.;Dellis,D.;Matthews,R.P.;Welton,T.;Hunt,P. A.J.Phys.Chem.B 2012,116,4921.doi:10.1021/jp209485y
(27) Feng,H.J.;Zhou,J.;Qian,Y.J.Chem.Phys.2011,135, 144501.doi:10.1063/1.3641486
(28) deAndrade,J.;Boes,E.S.;Stassen,H.J.Phys.Chem.B 2002, 106,13344.doi:10.1021/jp0216629
(29) Cornell,W.D.;Cieplak,P.;Bayly,C.I.;Gould,I.R.;Merz,K. M.;Ferguson,D.M.;Spellmeyer,D.C.;Fox,T.;Caldwell,J. W.;Kollman,P.A.J.Am.Chem.Soc.1995,117,5179.doi: 10.1021/ja00124a002
(30)Lyubartsev,A.P.;Laaksonen,A.Comput.Phys.Commun.2000, 128,565.doi:10.1016/S0010-4655(99)00529-9
(31) Iglesias-Otero,M.A.;Troncoso,J.;Carballo,E.J.Solut.Chem. 2007,36,1219.doi:10.1007/s10953-007-9186-6
(32) Gu,Z.;Brennecke,J.F.J.Chem.Eng.Data 2002,47(2),339. doi:10.1021/je010242u
(33) Mendez-Morales,T.;Carrete,J.;Garcia,M.;Cabeza,O.; Gallego,L.J.;Varela,L.M.J.Phys.Chem.B 2011,115,15313. doi:10.1021/jp209563b
(34) Liu,J.;Cao,D.P.;Zhang,L.Q.J.Phys.Chem.C 2008,112, 6653.doi:10.1021/jp800474t
(35)Abbott,A.P.ChemPhysChem 2005,6,2502.
(36)Noda,A.;Hayamizu,K.;Watanabe,M.J.Phys.Chem.B 2001, 105,4603.doi:10.1021/jp004132q
(37) Stoppa,A.;Hunger,J.;Buchner,R.J.Chem.Eng.Data 2009, 54,472.doi:10.1021/je800468h
(38) Zhang,S.J.;Lü,X.M.Ionic Liquids-from Fundamentals to Applications;Science Press:Beijing,2006;p 111.[张锁江,吕兴梅.离子液体-从基础研究到工业应用.北京:科学出版社,2006:111.]
(39) Laaksonen,L.J.Mol.Graph.1992,10,33.doi:10.1016/0263-7855(92)80007-Z
June 12,2012;Revised:August 24,2012;Published on Web:August 27,2012.
SimulationStudyoftheEffectofMethanolontheStructureandProperties of 1-Butyl-3-methylimidazolium Tetrafluoroborate Ionic Liquid
WANG Ding TIAN Guo-Cai*
(Faculty of Metallurgical and Energy Engineering,Kunming University of Science and Technology,Kunming 650093,P.R.China)
The microstructure and properties of 1-butyl-3-methylimidazolium tetrafluoroborate([BMIM] [BF4])/methanol mixtures with different amount-of-substance fractions for methanol(0.1-0.9)were studied by molecular dynamics(MD)simulations at 298.15 K and 0.1 MPa.The densities,radial distribution functions,coordination numbers,self-diffusion coefficients,viscosities,and conductivities of the systems were obtained.The simulated densities agreed with experimental values.As the methanol amount-ofsubstance fraction increased,the radial distribution functions of different components in the mixture showed regular changes,the interaction between the anion and cation and the viscosity decreased,and the conductivity and the self-diffusion coefficients increased.The spatial distribution functions obtained from the MD simulations were visualized to depict the microscopic structures of different components in the system.
Ionic liquid;1-Butyl-3-methylimidazolium tetrafluoroborate;Methanol;Molecular dynamics simulation;Microstructure;Physicochemical property
10.3866/PKU.WHXB201208271
∗Corresponding author.Email:tiangc01@gmail.com;Tel:+86-870-5162008.
The project was supported by the National Natural Science Foundation of China(50904031,51264021),Back-up Personnel Foundation ofAcademic and Technology Leaders of Yunnan Province,China(2011CI013),and Natural Science Foundation of Yunnan Province,China(2008E0049M).
国家自然科学基金(50904301,51264021),云南省中青年学术技术带头人后备人才(2011CI013)和云南省自然科学基金(2008E0049M)资助项目
O643;O645
猜你喜欢
工业水处理(2022年12期)2023-01-05
河北画报(2020年10期)2020-11-26
西南石油大学学报(自然科学版)(2018年6期)2018-12-26
英美文学研究论丛(2018年2期)2018-08-27
中国洗涤用品工业(2015年7期)2015-02-28
现代农业(2015年3期)2015-02-28
应用化工(2014年1期)2014-08-16
郑州大学学报(理学版)(2012年4期)2012-03-25
中国洗涤用品工业(2012年8期)2012-03-20
中国洗涤用品工业(2011年4期)2011-03-20