
鄂霍次克海冷泉沉积物真核生物多样性的初步研究
2012-10-23 03:01李富超栾锡武秦蕴珊
海洋科学 2012年5期
高 远, 李富超 姜 鹏 秦 松 栾锡武 秦蕴珊
(1. 中国科学院 海洋研究所, 山东 青岛 266071; 2. 中国科学院 研究生院, 北京 100049)
鄂霍次克海冷泉沉积物真核生物多样性的初步研究
高 远1,2, 李富超1, 姜 鹏1, 秦 松1, 栾锡武1, 秦蕴珊1
(1. 中国科学院 海洋研究所, 山东 青岛 266071; 2. 中国科学院 研究生院, 北京 100049)
从鄂霍次克海沉积物样品中提取总基因组DNA, 进行核糖体rRNA基因内转录间隔区(ITS)的扩增, 构建克隆文库并进行测序, 对该环境下真核生物的多样性进行了初步研究。结果发现, 该环境下的真核生物具有较高的多样性, 主要为 3大类, 真核藻类、真菌和微型浮游动物。藻类和微型浮游动物的组成与前人在该海域浮游生物生态学的研究相吻合, 而真菌群落具有较高的多样性, 这为该区域真核生物多样性、所参与的生物地球化学过程及生态学意义的研究奠定了基础。
冷泉沉积物; 核糖体rRNA基因内转录间隔区; 真核生物多样性
冷泉是指温度接近于海水, 而以高于周围水环境浓度的烃类化合物(主要为甲烷)、硫化物或二氧化碳为主要成分, 受地质构造或压力梯度作用渗出沉积物表层的流体[1]。海底冷泉作为一种极端环境具有独特的生物多样性和极高的生物密度[2], 该环境下的生物群落逐渐被发现和报道, 已成为地质学、生物学的研究热点, 为地球生物化学、生命演化和极端生命的研究提供了机遇。
通过环境样品基因序列的分析来研究环境生物多样性被广泛应用于原核微生物生态学研究, 并已成为不可或缺的研究手段[3]。然而该方法在真核生物生态学方面尚未被普遍采用。Heath[4]曾通过该方法对森林土壤环境样品中的真菌群落进行过研究, 其使用了真核生物通用的核糖体DNA内转录间隔区引物 ITS1/ITS4, 发现相对于传统的培养方法, 该方法发现了极为丰富的生物多样性, 而与 DGGE等指纹图谱方法相比能够提供前者所不能提供的完全的物种信息。Lai等[5]通过构建 ITS的克隆文库, 分析了南海甲烷水合物富集区的真菌群落多样性。转录间隔区是真核生物中分隔核糖体大亚基与小亚基rRNA基因的区域, 由于其进化速率较快, 可用于很多真菌、植物和藻类属和种水平上的识别和鉴定。目前对于该序列, 已经具有基因信息量很大的数据库[6], 这为该序列在多样性研究方面的应用提供了基础。
鄂霍次克海位于太平洋的西北角, 千岛岛弧的内侧, 海域面积仅次于中国的南海, 是西北太平洋大陆边缘中第二大边缘海, 其三面被高耸的山脉环绕, 这为该地区提供了大量有机及无机的沉降颗粒[7], 通过对海底沉积物中生物多样性的研究将对该海域浮游生物生态学提供佐证。同时由于鄂霍次克海的海底是一种典型的冷泉环境, 在该海域进行该类研究还将有助于对极端生命的探索。
1 材料与方法
1.1 采样
2006年5月由俄、韩、日、中四国共同组织的鄂霍次克海天然气水合物联合调查航次, 取得了海底表层沉积物的柱状样品。沉积物样品名: LV39-18H;采样深度: 715 m; 经纬度: 54°29.92′N, 144°16.53′E;由中国科学院海洋研究所海洋地质与环境重点实验室提供。
1.2 DNA的提取
通过FastDNA Spin Kit for Soil(MP Biomedicals,美国)试剂盒对该柱状沉积物 0~40 cm 的表层样品0.35 g进行总DNA的提取。
1.3 真核生物ITS片段的扩增
1.3.1 PCR反应体系及反应条件
使用 ITS通用引物 ITS1 (5′-TCCGTAGGTGAACCTGCGG)和 ITS4(5′-TCCTCCGCTTAT TGATATGC)[8], 使用实验方法1.2中提取的DNA作为模板。PCR反应体系如下: DNA模板 0.5 μL; ITS1引物(10 μmol/L)1 μL, ITS4 引物(10 μmol/L)1 μL; 2×MasterMix Taq酶 12.5 μL (天根生化科技有限公司, 中国);Mili Q水补至 25μL。使用 Biometra Tprofessional PCR仪(德国), 反应条件如下: 94°C预变性 6 min;94°C 50 s, 50°C 50 s, 72°C 70 s, 循环 30 次; 72°C 延伸10 min。
1.3.2 PCR产物的检测
使用1%琼脂糖凝胶对PCR产物进行电泳, 溴化乙锭染色后于紫外凝胶成像分析系统(Bio-Rad, 美国)下进行检测。
1.4 构建克隆文库
使用 DNA琼脂糖凝胶回收试剂盒(Bioteke, 中国)对 PCR 产物进行回收。与克隆载体pMD18-T(TaKaRa, 日本)连接并转化到Escherichia coliTop10(Invitrogen, 加拿大)细胞中, 于 37°C 恒温培养过夜。挑取48个单菌落, 送到上海美吉生物医药科技有限公司进行序列测定。
1.5 序列分析
通过 BLAST软件从 GenBank获得与本实验所得到序列亲缘关系最近的序列。使用 CLUSTAL W软件[9]进行多序列比对。根据97%的分类标准, 将所得的序列使用 DOTUR软件[10]进行系统发育类型的归类。使用DOTUR对文库进行各统计学指数的分析:稀有度曲线作为判断文库系统发育类型丰富度的参考; Shannon多样性指数用来评价系统发育类型多样性; Chao1 丰富度指数用来推断样品中物种总数。
2 结果
2.1 对所得序列的统计学分析
对 48个单菌落进行测序, 去除不成功的序列,共获得 38条序列, 通过 DOTUR软件分析, 所有序列根据 97%的分类标准被划分为 19个系统发育类型。
通过 BLAST检索, 发现所有的 38条序列主要来自3大类, 分别为藻类(19条序列)、真菌(8条序列)和浮游动物(11条序列), 分别占克隆总数的 50%,21.1%和28.9%。3大类群中具体门类所占比例如图1所示, 对各大类群的统计分析如表1所示, 各大类群在文库中的稀有度曲线如图2所示。
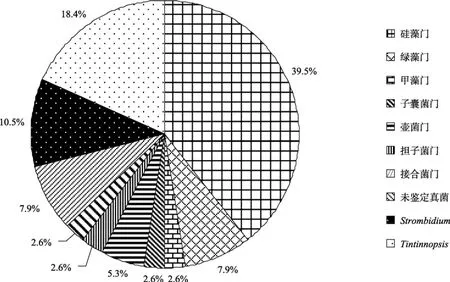
图1 各真核生物类群占ITS克隆文库的比例Fig. 1 Proportion of each eucaryotic group in the ITS clone library藻类用交叉线表示, 真菌用直线表示, 浮游动物用点表示Algae represented by crossed lines, fungi represented by straight lines and zooplankton represented by dots

表1 对真核生物ITS文库的统计学分析Tab. 1 Statistical analysis of ITS library

图2 ITS克隆文库各类群的稀有度曲线Fig. 2 Rarefaction curve of each group in the ITS clone library
具体来说, 根据Shannon多样性指数显示, 真菌的多样性最高, 达到2.08, 浮游动物多样性最低, 为1.04。Chao1指数也表现出相似的趋势, 真菌的Chao1指数达到36, 而浮游动物该指数仅为3。就整个ITS文库来说, Shannon指数和Chao1指数分别达到2.56和38.5。
从稀有度曲线可以看出, 随着克隆抽样次数的增加, 真菌系统发育类型数目表现出同步增长趋势,即完全未达到饱和。与此相反, 浮游动物的系统发育类型基本达到饱和并最终稳定在 3左右, 而藻类表现出部分饱和。
2.2 真核生物多样性
2.2.1 藻类多样性
属于藻类的19条序列可被划分为8个系统发育类型。具体来说, 5个系统发育类型属于硅藻, 另有2个和 1个系统发育类型分别属于绿藻和甲藻。其中属于3个系统发育类型的12条序列与硅藻角毛藻属(Chaetoceros)的亲缘关系最高(相似度分别为 97%、92%和 93%), 该属为藻类中的优势类群, 占藻类总克隆数目的63.2%。另有属于2个系统发育类型的3条序列与绿藻门的海洋绿色微藻Koliella spiculiformis的亲缘关系最高(相似度分别91%和90%)。此外还发现了 4条序列分别与硅藻门双环海链藻属(Thalassiosira, 一条序列, 相似度 85%)和短缝藻属(Eunotia, 两条序列, 相似度87%)以及甲藻门裸甲藻属(Gymnodinium, 一条序列, 相似度 84%)有较低的相似度。
2.2.2 真菌多样性
属于真菌的所有 8条序列分别属于不同的系统发育类型, 即共可划分为8个系统发育类型。它们分别来自壶菌门、担子菌门、接合菌门和子囊菌门, 另有3条序列分别与环境样品中获得的ITS序列亲缘关系最近, 目前尚无法具体鉴定。其中来自子囊菌门的序列与海洋酵母菌梅奇酵母属(Metschnikowia)有93%的相似度。
2.2.3 浮游动物多样性
属于浮游动物的11条序列可被划分为3个系统发育类型, 经过BLAST检索发现, 所有3个系统发育类型都属于 20~200 μm 微型浮游动物的纤毛虫,其中两个系统发育类型的序列与无壳纤毛虫的Strombidium具有较高同源性(相似度为98%和96%),而另一系统发育类型的序列与砂壳纤毛虫的Tintinnopsis具有较高同源性(相似度为97%)。
3 讨论
从鄂霍次克海沉积物样品中提取总基因组 DNA,进行 ITS基因的扩增并构建文库对该环境下真核生物的多样性进行了研究。
统计学分析特别是 Shannon指数提示, 在该冷泉沉积物环境中真菌具有极高的多样性, 而浮游动物的多样性最低。根据饱和曲线显示出相似结果, 由于挑取的克隆数较少, 真菌多样性曲线完全未达到饱和, 但是浮游动物多样性已基本达到饱和。基于以上多样性分析, 我们建议使用真菌特异的 ITS引物或增加测序克隆数目, 以对真菌类群的多样性进行进一步研究。
本文中发现的藻类序列大部分来自能引起赤潮的浮游藻类。其中角毛藻在许多文献中记载为鄂霍次克海域最主要的浮游藻类[11], 本文的结果也与前人对浮游生态学的研究相吻合。有趣的是, 在我们对该沉积物 16S rRNA基因构建的克隆文库中也发现了占该文库较高比例的角毛藻属叶绿体 16S rRNA基因(数据未发表), 且此类群在沉积年代数万年的深度仍有发现, 因此推测本研究发现的这些序列很可能来自这类浮游硅藻产生的随沉积作用进入沉积物的休眠细胞。此外与赤潮藻类硅藻门双环海链藻属和甲藻门裸甲藻属有一定亲缘关系的序列在本文中也有发现。总体来说, 硅藻在所有这些赤潮藻类中占绝对优势, 这可能是因为与甲藻等其他赤潮藻类相比, 硅藻主要采取r-对策, 即通过产生大量的有数月乃至几年生活力的休止细胞, 能够迅速适应适宜环境。在海底沉积物中, 硅藻休止细胞数目占有绝对优势的现象也屡次通过常规生态学方法检测到并被报道[12]。
本文中发现的真菌多样性大大高于其余两大类群, 这提示在海洋沉积物环境中的真菌群落有可能为我们提供丰富的药源和基因资源, 比如梅奇酵母属是重要的海洋酵母菌, 已有对其酸性蛋白酶基因进行开发的相关报道[13]。因此对海洋真菌的进一步研究将有着重要的应用和经济价值。
无壳纤毛虫和沙壳纤毛虫是两类主要的 20~200 μm微型浮游动物, 在世界许多海域具有较高的生物量[14]。本研究发现的上述两类浮游动物的序列, 分别属于Strombidium和Tintinnopsis。前者为鄂霍次克海中一种优势浮游动物[15], 而后者在鄂霍次克海的分布也早在1932年就由Hada[16]报道过。
4 结论
本文的序列大部分来源于鄂霍次克海中的浮游藻类和浮游动物, 通过沉降作用到达沉积物中, 本文的研究在一定程度上为浮游生态学的研究提供了佐证。真菌是本研究中发现的 3大类群中多样性最高的类群, 对该类群的进一步研究将有重要的科研和经济价值。此外, 本研究也证明通过对ITS序列进行扩增进而构建克隆文库的分子生物学方法确实能够发现较高的多样性并方便进行有效的统计学分析,这为今后对该区域真核生物特别是真菌的多样性、所参与的生物地球化学过程及生态学意义的研究奠定了基础。
[1] Reed A J, Lutz R A, Vetriani C. Vertical distribution and diversity of bacteria and archaea in sulfide and methane-rich cold seep sediments located at the base of the Florida Escarpment[J]. Extremophiles, 2006, 10:199-211.
[2] Levin L A. Ecology of cold seep sediments: interactions of fauna with flow, chemistry and microbes[J]. Oceanogr Mar Bio, 2005, 43: 1-46.
[3] DeLong E F, Pace N R. Environmental diversity of bacteria and archaea[J]. Syst Biol, 2001, 50: 470-478.
[4] Heath E O, Jeri L P, Jason A J, et al. Fungal community analysis by large-scale sequencing of environmental samples[J]. Appl Environ Microbiol, 2005, 71:5544-5550.
[5] Lai X, Cao L, Tan H, et al. Fungal communities from methane hydrate-bearing deep-sea marine sediments in South China Sea[J]. ISME J, 2007, 1: 756-762.
[6] Vilgalys R. Taxonomic misidentification in public DNA databases[J]. New Phytol, 2003, 160: 4-5.
[7] 栾锡武, 赵克斌, Obzhirov A, 等. 鄂霍次克海浅表层天然气水合物的勘查识别和基本特征[J]. 中国科学,2008, 38: 99-107.
[8] White T J, Bruns T D, Lee S, et al. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics[C]//Innis M A, Gelfand D H, Sninsky J J,et al. PCR protocols: a guide to methods and applications. New York, NY: Academic Press Inc., 1990:315-324.
[9] Thompson J D, Higgins D G, Gibson T J. CLUSTAL W:improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice[J].Nucleic Acids Res, 1994, 22: 4673-4680.
[10] Schloss P D, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness[J]. Appl Environ Microbiol, 2005, 71: 1501-1506.
[11] Zakharkov S P, Selina M S, Vanin N S, et al. Phytoplankton characteristics and hydrological conditions in the western part of the sea of Okhotsk in the spring of 1999 and 2000 based on expeditionary and satellite data[J]. Mar Biol, 2007, 47: 559-570.
[12] 谢文玲, 高亚辉. 海洋浮游植物休眠期的生态学研究[J].厦门大学学报(自然科学版), 2006, 45: 240-244.
[13] Li J, Chi Z, Liu Z, et al. Cloning and characterization of a novel aspartic protease gene from marine-derived Metschnikowia reukaufii and its expression in E. coli[J].Appl Biochem Biotech, 2009, 159: 119-132.
[14] 张武昌, 肖天, 王荣. 海洋微型浮游动物的丰度和生物量[J]. 生态学报, 2001, 21: 1893-1908.
[15] Sorokin Y, Sorokin P. Production in the sea of Okhotsk[J]. J Plankton Res, 1999, 21: 201-230.
[16] Hada Y. Tintinnoinea from the sea of Okhotsk and its neighborhood[J]. J Fac Sci Hokkaido Univ, 1932, 2:37-59.
Study of eukaryotic diversity in cold seep sediment of the Okhotsk Sea
GAO Yuan1,2, LI Fu-chao1, JIANG Peng1, QIN Song1, LUAN Xi-wu1,QIN Yun-shan1
(1. Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China; 2. Graduate School of the Chinese Academy of Sciences, Beijing 100049, China)
May,11,2011
cold seep sediments; internal transcribed spacers; eukaryotic diversity
The eukaryotic diversity in the sediment sampled from the Okhotsk Sea was evaluated using direct isolation of internal transcribed spacer (ITS) rRNA genes by PCR and sequencing of cloned fragments. It is shown from the results that the eukaryotic community exhibited high diversity and all of the sequences were from three groups:algae, fungus and microzooplanktons. The constructions of algal and microzooplanktonic communities were consistent with previous ecological research results in this area. Interestingly, we found that the fungal community showed relatively high diversity, which provided clues for further research.
X17
A
1000-3096(2012)05-0029-05
2011-05-11;
2011-07-23
中国科学院知识创新工程项目——深海极端环境微生物生命过程研究(KZCX2-YW-JC201), 深海沉积物中甲烷代谢相关微生物群落的元基因组学探索性研究(KSCX-2-YW-G-022, 2007-1); 中国大洋协会项目——深部生物圈微生物活动在沉积物和间隙水中的地球化学记录研究(DYXM-115-02-2-17)
高远(1983-), 男, 山东青岛人, 硕士研究生, 主要从事深海微生物生态学研究, E-mail: gy.james@163.com; 秦松, 通信作者,男, 研究员, 博士生导师, 主要从事海洋生物工程研究, E-mail:sqin@ms.qdio.ac.cn
(本文编辑:张培新)
猜你喜欢
当代水产(2021年8期)2021-11-04
现代畜牧科技(2021年8期)2021-10-13
昆明医科大学学报(2021年5期)2021-07-22
猪业科学(2021年3期)2021-05-21
军事文摘(2020年20期)2020-11-16
幽默大师(2020年10期)2020-11-10
中华诗词(2019年1期)2019-11-14
今日农业(2019年10期)2019-01-04
生命科学研究(2018年1期)2018-05-29
猪业科学(2018年4期)2018-05-19
