
绿脓杆菌分子分型方法研究进展
2012-09-17 14:58岳秉飞贺争鸣
中国比较医学杂志 2012年8期
邢 进,岳秉飞,贺争鸣
(中国食品药品检定研究院 实验动物资源研究所,北京 100050)
绿脓杆菌分子分型方法研究进展
邢 进,岳秉飞,贺争鸣
(中国食品药品检定研究院 实验动物资源研究所,北京 100050)
绿脓杆菌是一种常见的人畜共患机会致病菌,广泛存在于自然界,是造成实验动物污染和医院内感染的重要病原菌之一。分子分型方法是病原菌流行病学分析的重要手段,对于确定感染来源和途径,检测交叉污染和流行菌株方面非常有效。本文主要对绿脓杆菌分子分型方法的研究进展进行综述。
绿脓杆菌;分子分型方法;动物,实验
对病原菌的特征分析及准确分型,了解各种不同来源的菌株是否具有相同的起源,在流行病学分析中必不可少。细菌分型包括表型分型和分子分型两种方法,对查找感染源和感染途径、鉴别地方性菌株和流行性菌株、预防交叉感染非常重要。传统的表型方法只靠表型不足以区别相同基因型的菌株。分子分型方法经过十多年的发展,已具备很好的稳定性和分辨力,成为了解细菌感染的流行病学特征必不可少的技术。
绿脓杆菌,又称铜绿假单胞菌(Pseudomonas aeruginosa,PA)是一种常见的人畜共患机会致病菌,广泛存在于自然界。该菌也是医院内感染的重要病原菌之一[1],在临床上是导致囊性纤维化患者(CF)、烧伤患者和其他免疫功能低下患者院内感染的主要病原[2,3]。由于其自身的天然耐药性以及广谱抗生素的广泛使用,使得多重耐药绿脓杆菌感染的情况日益严重[4]。在动物中,绿脓杆菌同样是作为一种机会致病菌存在于体内,呼吸道和肠道内均可分离得到。一般情况下并不引起动物发病,当动物免疫力低下时,才会引起动物发病,甚至死亡。
建立经济可靠的分型系统能够更好的对绿脓杆菌进行流行病学分析,各种分子分型方法在绿脓杆菌的流行病学研究中均有应用[5-8],弥补了表型研究的不足。本文通过比较绿脓杆菌各种分型方法的特点,希望能够帮助我们选择方便、快捷、经济和可靠的分型方法,对绿脓杆菌进行流行病学分析,设计更合理的感染控制策略。
1 以PCR为基础的分型方法
1.1 随机扩增多态性(random amplification of polymorphic DNA,RAPD)指纹图谱
RAPD技术是1990年由Williams等人建立的一种分析基因多态性的分型方法[7],是在PCR基础上发展起来的一种分子生物学技术。RAPD-PCR通常使用的是9~10 bp的单一随机引物,对目标基因组进行随机扩增,随机扩增出多条非特异性条带,不同的菌株所得到的条带是不同的。保持RAPD分析的可靠性和重复性,适合的退火温度和模板DNA的质量至关重要。PCR的其他条件,如Mg2+浓度、引物浓度和模板浓度等都需要合理的优化才能得到理想的指纹图谱[8]。在菌株遗传背景不清楚的情况下,RAPD-PCR仅通过几微克的模板DNA就可以对该种微生物进行分型研究。
临床上,RAPD-PCR与血清学、形态学等方法结合,能够很好的揭示绿脓杆菌的流行病学特征[9]。但是RAPD的缺点是由于很低的退火温度,反应体系细微的变化就会导致RAPD-PCR图谱结果的变化,重复性很差,而且缺乏标准化,不同实验室之间的绿脓杆菌PAPD结果无法在统一的平台上进行比较。由绿脓杆菌的分型结果表明,RAPD-PCR的分辨力优于RFLP和核糖体分型,但低于REP-PCR[10]。
1.2 扩增片段长度多态性(amplified fragment length polymorphism,AFLP)
AFLP最先由荷兰科学家Zebean和Vos在1992年发明的检测DNA多态性的一种方法[11],最初用于植物基因组的研究。其基本原理是利用1~3种限制性内切酶切割目的基因组DNA,通常使用2种内切酶,一种识别高频位点,一种识别低频位点,EcoR I和Mse I是最常用的两种酶。所得的扩增片段用聚丙烯酰胺凝胶电泳进行检测。若目的基因组酶切片段内的序列发生改变,或者酶切位点发生碱基突变等情况,最后的酶切片段会随之改变,PCR扩增片段的电泳图谱多态性也会相应变化。Pirnay等人用AFLP结合血清学、药敏等方法,对比利时河流内的绿脓杆菌多样性和相似性进行了研究,发现该流域具有独特的流行株[6]。AFLP能够很好的用于绿脓杆菌院内感染的流行病学研究,进行实时的感染暴发监测,能有效的揭示耐药菌株的来源、感染途径和分布特点,有助于感染的控制[12]。AFLP与其他分型方法相比,分型能力和分辨力取决于限制性内切酶和识别碱基的合理选择。Putignani等[13]对ICU病房内患者下呼吸道绿脓杆菌分离株的基因型分析中表明AFLP与rep-PCR的分型能力相当。Naze对新生儿病房绿脓感染的研究中显示AFLP的分型能力弱于 MLVA[7],而在有些情况下优于 PFGE[14]。
1.3 重复序列PCR(repetitive element sequencebased PCR,rep-PCR)
rep-PCR 是1991年由 Versalovic等[15]建立的一种细菌基因组指纹分析方法,由RAPD-PCR方法发展而来,通过扩增细菌基因组中高度保守重复序列,分析基因型间的差异[16]。rep-PCR根据细菌基因组中广泛分布的短重复序列的不同,又分为基因外重复回文序列(repetitive extragenic palindromic,REP)PCR、肠杆菌基因间重复一致序列(enterobacterial repetitive intergenic consensus,ERIC)PCR和 BOX-PCR。2004年朱琴应用ERIC-PCR对56株耐亚胺培南绿脓杆菌分离株进行了聚类分析,结果将56株耐药株分出了33 个型别[17]。Syrmis 等[18]人应用ERIC-PCR 和BOX-PCR方法对囊性纤维化患者中分离的绿脓杆菌进行了分型研究,并与PFGE方法进行了对比,结果显示两种rep-PCR方法的分型能力都非常理想。此外,Olive等人应用REP-PCR和ERIC-PCR方法的分型研究表明,其分辨力高于RAPD-PCR和核糖分型方法[19]。国内应用REP-PCR方法,对烧伤病房和肝移植中的绿脓杆菌分离株进行分型研究,为临床用药提供了依据[20-21]。Kidd等用不同方法对囊性纤维化患者(CF)中分离到的绿脓杆菌进行的分型研究表明,ERIC-PCR 的分辨力与 MLST、PFGE 相比略低[4]。BOX-PCR方法对绿脓杆菌分型的应用较少。总之rep-PCR中的上述三种主要方法对于绿脓杆菌的分型,在分辨率和重复性,效果相当。与其它分型方法相比,优于RFLP、核糖体分型,比PFGE方法的分辨力略低,但相对操作简便,经济,可实现自动化分型,在绿脓杆菌流行病学监测中发挥重要作用。rep-PCR的主要缺点是所有的遗传多态性在细菌基因组的特定位置,这必然会导致其在分辨力上的局限性。
1.4 多位点可变数目串联重复序列分析(multiplelocus variable-number tandem-repeat analysis,MLVA)
MLVA方法来源于微卫星的基因标记技术,是对基因组中可变数目重复序列(variable-number tandem-repeat,VNTR)的分析方法。针对每个重复序列区段涉及特异性的PCR引物,每个位点内的重复序列数目一般通过估计此位点的PCR片段大小来确定。一般细菌基因组中可用于分型的位点在20个左右,比如肠炎沙门氏菌、炭疽芽孢杆菌、鼠疫耶尔森氏菌等。
绿脓杆菌的 MLVA分型研究中,Onteniente等用绿脓杆菌PAO1株的全基因组序列为参考,建立了7个 VNTR位点的 MLVA方法[22]。Vu-Thien等人随后建立了15个VNTR位点的MLVA方法,这种位点的组合分型更加有效、简便和快捷。目前MLVA对绿脓杆菌分型的研究主要就是采用的这个方法,具有很好的多态性,这 15个位点分别是ms77、ms127、ms142、ms172、ms211、ms212、ms213、ms214、ms215、ms216、ms217、ms222、ms223[23-24]。而Naze等[3]人建立了仅采用其中一个位点的MLVA方法,在PCR产物的电泳处理上,不是采用普通的琼脂糖凝胶电泳,而是采用聚丙烯酰胺凝胶电泳,这种方法使MLVA的分辨力提高了14%。van Mansfeld等人在Vu-Thien的研究基础上,建立并优化了9个位点的分型方法,与PFGE和MLST两种方法在分辨力、经济性和便捷性的综合比较是最佳的分型方案[25]。总之,MLVA方法的优点是不同绿脓杆菌的VNTR的数目能够数字化,通过互联网上的数据库可以进行比对,菌株的异同一目了然。MLVA方法的关键是对各VNTR片段的大小进行确定,对重复序列长度太小的VNTR位点(小于15nt),必须要进行DNA测序才能够得出其重复序列数[27]。虽然有自动化的设备和软件可以对这些片段大小进行分析,但是价格很高。目前,MLVA只适用于已知全基因组序列的细菌,因此其应用受到了限制。
1.5 多位点序列分析(multilocus sequencing typing,MLST)
MLST方法作为一种新的分型技术,源于多位点酶电泳(multilocus enzyme electrophoresis,MLEE),最初是由 Maiden MC等人在1998年建立并应用于对脑膜炎奈瑟菌的分型[26]。它是使用多个(7个以上)400~500 bp大小的管家基因位点的序列型(ST)来描述每一株细菌的不同[27]。Curran等人最先使用 7个管家基因(acsA、aroE、guaA、mutL、nuoD、ppsA和trpE)对绿脓杆菌的 ST进行分析,证明此方法能够很好的对绿脓杆菌临床分离株进行分型[28-29]。Johnson等人用 MLST和 PFGE 方法对绿脓杆菌进行分型比较,结果MLST的分辨力略高于 PFGE[30]。
MLST是一种操作简便的分子分型方法,其过程只是直接对生物样品中的DNA片段进行PCR扩增,然后对所得到的管家基因片段进行测序[26]。测序所得到的ST结果可以在互联网上的MLST数据库进行对比[31]。然而MLST的不足也显而易见,虽然其适合于菌株间的进化和相关性分析,但是对于短期内的临床分离株分型在分辨力上存在不足,特别是与PFGE和MLVA方法的比较中证明了这一点[25][30]。在几种主流分型方法中,即便不算仪器设备和分析工具的成本,MLST所需费用也是最高的[25],因此对绿脓杆菌临床应用并不是很多。
2 以限制性酶切为基础的分型方法
2.1 核糖体分型(ribotyping)
Ribotryping是一个基于不同细菌16S和23S rRNA的高度保守序列建立的分子分型方法[10],通过限制性核酸内切酶对细菌基因组DNA的进行酶切,利用凝胶电泳分离不同长度的片段。再用序列标记的DNA探针对酶切片段进行杂交,该探针包含编码高度保守16S rRNA和23S rRNA的DNA片段及间隔序列。由于一个生物的基因组中通常有多个核糖体基因,分别存在于不同长度的酶切片段中,因此核糖体分型可以得到类似指纹的结果,对细菌进行鉴定和分型。该方法可以通过选择两种酶的平行实验,对一种内切酶不能识别的区域进行酶切,增加对菌株的分辨力。研究发现由于核糖体DNA限制性内切位点的变异非常慢,菌株的变异不易被检测到,以至于核糖体分型图谱的变化很小,可用于评价分离株之间的同源关系[32]。核糖体分型可用于对临床绿脓杆菌耐药菌株的相关性研究,不过与PFGE和ERIC-PCR等方法的比较研究显示出了其分型能力和分辨力上的不足[10]。不过通过与血清分型等其它表型方法结合使用可以达到不错的效果[33]。核糖体分型最大的优势是实现了自动化,RiboPrinter全自动微生物基因指纹鉴定系统使研究人员在一天内就可得到细菌的rDNA指纹杂交图谱。
2.2 限制性酶切片段长度多态性(restriction fragment length polymorphism,RFLP)
RFLP源于生物基因组DNA的自然变异,RFLP作为第一代DNA分子遗传标记技术,极大的推动了人类DNA多态性的研究,现在已广泛应用于用于基因组遗传图谱构建、基因定位以及生物进化和分类的研究。通过对分离出的DNA片段转印或者其他方法分离,然后通过标记的探针对相应的基因进行检测[19],构建分子图谱。RFLP就能够检测出碱基插入、缺失、重排或点突变所引起的差异,从而比较不同菌株间DNA水平的差异。一般常用的限制性内切酶有 Hind III,BamH I,EcoR,EcoR V,Xba I等。RFLP对靶序列的浓度和质量要求很高,低含量、不纯的基因组DNA会严重影响RFLP分析结果。探针可以提高RFLP的分辨力,绿脓杆菌常用外毒素exoA基因探针用于分子流行病学分析[33]。由于重复性和分型能力上的不足单纯的RFLP方法已经很少应用于菌株分型。有比较表明,RFLP远不如RAPDPCR和PFGE,所以产生了PCR-RFLP技术,可以更准确的检测绿脓杆菌流行株的基因突变情况[6]。
2.3 脉冲场凝胶电泳(pulsed-fieldgel electrophoresis,PFGE)
PFGE是一种脉冲场凝胶电泳技术,使用相应的限制性内切酶对细菌基因组DNA上的酶切位点进行识别切割,产生若干10~800 kb的 DNA大片段,通过这些酶切片段的多态性分析菌株之间的相似性。1984年,Schwartz和Cantor报道了脉冲场电泳的方法[34],对至少40种病原菌进行了 PFGE分型试验,其中包括绿脓杆菌,解决了分离大片段DNA 难以分离的难题[27]。
PFGE是目前公认的用于绿脓杆菌及其他病原菌分子分型和溯源研究的“金标准”。PFGE所产生的酶切图谱与 RAPD、MLVA、MLST、核糖体分型和RFLP等大多数分子分型方法相比是更有效的分型工具。Hector试验了65种限制性内切酶用于对绿脓杆菌的酶切效果,结果证实SPE I是最佳的选择。不仅所有的酶切片段大于200 kbp,而且片段数目在30条左右,数目适中。到目前为止,绿脓杆菌的PFGE分型方法已经非常成熟,从使用的酶、酶切时间、电泳条件等都得到了优化,Romling等人的研究使PFGE中DNA降解的情况得以控制,对绿脓杆菌的分型能力达到了100%[35]。1996年,美国疾病预防控制中心以PFGE技术为基础建立了PulseNet细菌分子分型网络,我国疾病预防控制中心(CDC)也已加入这个网络(PulseNet China),此网络目前提供PFGE标准操作规程的下载,包括沙门氏菌、大肠杆菌、空肠弯曲菌食源性病原菌的标准PFGE方法,用以提高对食源性疾病的快速检测和溯源能力。实验人员对于电泳结果的理解和分析各不相同,对结果的分析就会出现很大差异,因此 Tenover等人经过多年的研究提出了 PFGE的解释标准[36]。作为经典的分子分型方法,PFGE已被广泛的应用于各种细菌的流行病学研究中,适用于菌株间基因同源性的分析[30]。不过 PFGE的操作相对复杂、费时、检测通量小[19,37],是制约其应用的不利因素。
3 结论
绿脓杆菌容易发生变异,很难用表型方法分辨菌株间的异同,甚至导致错误的结果[10-27]。分子分型方法能够弥补表型分型的不足,理想的分型方法需要具备以下的特征:①100%的分型能力,即能够将所有受试菌株全部分型;②高分辨能力,即将同源关系远的菌株分开,而将同一克隆或遗传相近的菌株聚类到一起;③良好的重复性;④操作简便、节约时间,并且费用低。由此可见,目前还没有完美的分型方法,每种分子分型方法包含不同分子生物学技术组成(表1),各有优点(表2)。同时,所有的分子分型方法都存在一个很大的局限性,即不能够对当前的流行株进行实时分析[38]。

表1 不同分型方法涉及的技术Tab.1 Techniques used in different molecular typing
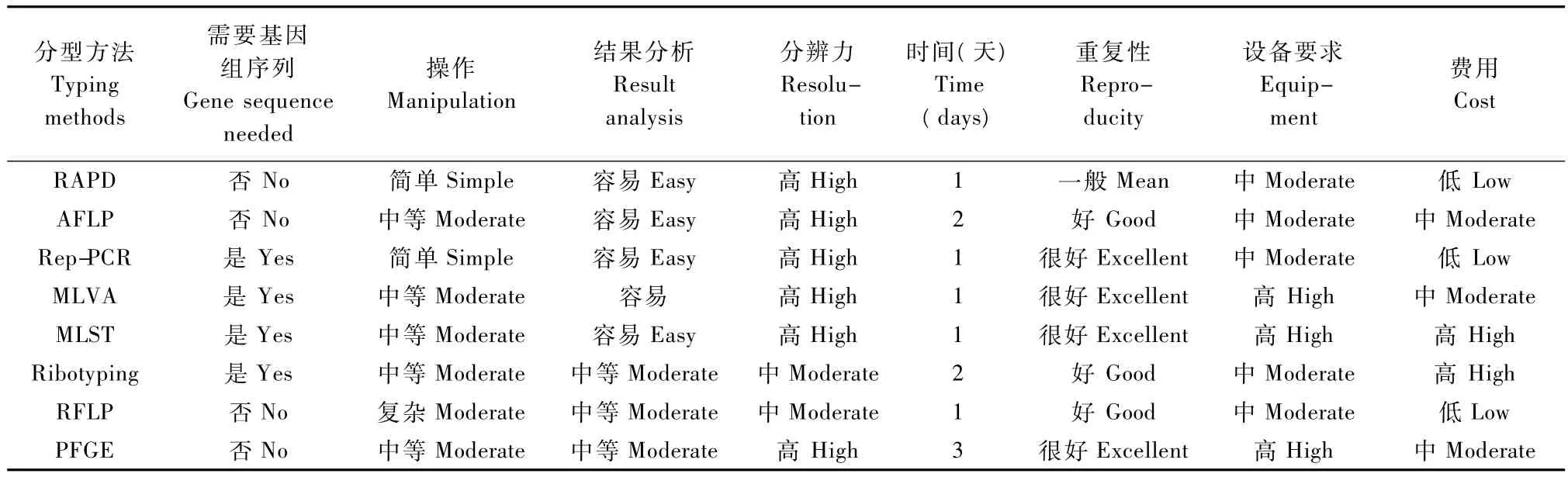
表2 不同分型方法之间特点比较Tab.2 Comparison of different typing methods
总之,低成本化、自动化、标准化的分子分型方法是细菌分型研究的发展方向。随着科学技术的发展,现有技术会不断得到完善,同时又会有新的方法不断涌现。分型方法是我们在微生物学、遗传学、流行病学等一系列研究中的工具,选择适合而合理的方法,有效监测微生物种群的分子流行病学变化,才能达到研究预期和最终目的。
[1]Goldberg JB.Why is Pseudomonas aeruginosa a pathogen?[J].F1000 Biology Reports.2010,2:29.
[2]Livermore DM.Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa:our worst nightmare?[J].Clin Infect Dis,2002,34:634-640.
[3]Naze F,Jouen E,Randriamahazo RT,et al.Pseudomonas aeruginosa outbreak linked to mineral water bottles in a neonatal intensive care unit:fast typing by use of high-resolution melting analysis of a variable-number tandem-repeat locus[J].J Clin Microbiol,2010,48(9):3146-3152.
[4]Kidd TJ,Grimwood K,Ramsay KA et al.Comparison of three molecular techniques for typing Pseudomonas aeruginosa isolates in sputum samples from patients with cystic fibrosis[J].J Clin Microbiol,2011,49(1):263-268.
[5]Wolska K,Kot B,Jakubczak A,et al.BOX-PCR is an adequate tool for typing of clinical Pseudomonas aeruginosa isolates[J].Folia Histochem Cytobiol,2011,49(4):734-738.
[6]Salimi H,Owlia P,Yakhchali,B,et al.Characterization of Pseudomonas aeruginosa in burn patients using PCR-restriction fragment length polymorphism and random amplified polymorphic DNA Analysis[J].Iran J Med Sci,2010,35(3):236-241.
[7]Williams JG,Kubelik AR,Livak KJ,et al.DNA polymorphisms amplified by arbitrary primers are useful as genetic markers[J].Nucleic Acids Res,1990,18(22):6531-6535.
[8]叶明亮,王全力.铜绿假单胞菌随机扩增多态性DNA分析中反应条件优化方案的探讨[J].军事医学科学院院刊,2001,25(3):186-189.
[9]Hafiane A,Ravaoarinoro M.Characterization of Pseudomonas aeruginosa strains isolated from cystic fibrosis patients by different typing methods[J].Pathol Biol,2011,59:e109-e114.
[10]Wu F, Della-Latta P. Moleculartyping strategies. Semin Perinatol,2002,26:357-366.
[11]Vos P,Hogers R,Bleeker M,et al.AFLP:a new concept for DNA fingerprinting[M].Nucleic Acids Res,1995,21:4407-4414.
[12]Fanci R,Bartolozzi B,Sergi S,at al.Molecular epidemiological investigation of an outbreak of Pseudomonas aeruginosa infection in an SCT unit[J].Bone Marrow Transplant,2009,43:335-338.
[13]Putignani L,Sessa R,Petrucca A,et al.Genotyping of different Pseudomonas aeruginosa morphotypes arising from the lower respiratory tract of a patient taken to an intensive care unit[J].Int J Immunopathol Pharmacol,2008,21(4):941-947.
[14]Speijer H.,Savelkoul PH,Bonten MJ,et al.Application of different genotyping methods for Pseudomonas aeruginosa in a setting of endemicity in an intensive care unit[J].J Clin Microbiol,1999.37:3654-3661.
[15]Versalovic J,Koeuth T,Lupski JR.Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes[J].Nucleic Acids Res,1991,19:6823-6831.
[16]Hulton CSJ,Higgins CF,Sharp PM.ERIC sequence:a novel family of repetitive elements in the genomes of Escherichia coli,Salmonella typhimurium and other enterobacteria[J].Molec Microbiol,1991,5(4):825-834.
[17]朱琴,彭少华.铜绿假单胞菌耐亚胺培南现状与分子水平研究[D].武汉大学,2004,24.
[18]Syrmis MW,O’Carroll MR,Sloots TP,et al.Rapid genotyping of Pseudomonas aeruginosa isolates harboured by adult and paediatric patients with cystic fibrosis using repetitive-element based PCR assays[J]. J Med Microbiol,2004,53:1089-1096.
[19]Olive DM,Bean P.Principles and applications of methods for DNA based typing of microbial organisms. J Clin Microbiol,1999,37:1661-1669.
[20]孙珍,向军,宋菲,等.烧伤病房铜绿假单胞菌的流行性分析[J].中华医院感染学杂志,2011,21(18):3774-3776.
[21]李浩,顾雁,邢同海,等.肝移植受者中多重耐药革兰阴性菌的检测和同源性分析[J].中国感染与化疗杂志,2011,11(6):457-462.
[22]Onteniente L,Brisse S,Tassios P.T,et al.Evaluation of the polymorphisms associated with tandem repeats for Pseudomonas aeruginosa strain typing[J].J Clin Microbiol,2003,41(11):4991-4997.
[23]Vu-Thien H,CorbineauG,Hormigos K.Multiple-locus variable-number tandem-repeat analysis for longitudinal survey of sources of Pseudomonas aeruginosa infection in cystic fibrosis patients[J].J Clin Microbiol,2007,45(10):3175-3183.
[24]http://minisatellites u-psud fr/MLVAnet/[OL].2012-2-9.
[25]van Mansfeld R,Jongerden I,Bootsma M,et al.The population genetics of Pseudomonas aeruginosa isolates from different patient populations exhibits high-level host specificity[J].Plos One,2010,5(10):e13482.
[26]Maiden MC,Bygraves JA,Feil E,et al.Multilocus sequence typing:a portable approach to the identification of clones within populations of pathogenic microorganisms[J].Proc Natl Acad Sci,1998,95(6):3140-3145.
[27]Singh A,Goering RV,Simjee S,et al.Application of molecular techniques to the study of hospital infection[J].Clin Microbiol Rev,2006,19:512-530.
[28]Curran B,Jonas D,Grundman H,et al.Development of a multilocus sequence typing scheme for the opportunistic pathogen Pseudomonas aeruginosa[J].J Clin Microbiol,2004,42:5644-5649.
[29]Vernez I,Hauser P,Bernasconi MV,et al.Population genetic analysis of Pseudomonas aeruginosa using multilocus sequence typing[J].FEMS Immunol Med Microbiol,2005,43:29-35.
[30]Johnson JK,Arduino SM,Stine OC,et al.Multilocus sequence typing compared to pulsed-field gel electrophoresis for molecular typing of Pseudomonas aeruginosa[J].J Clin Microbiol,2007,45(11):3707-3712.
[31]Keith Jolley.http://pubmlst.org/paeruginosa[OL].University of Oxford,UK.2012-2-11.
[32]Grattard F, Pozzeto B, Ros A, et al. Differentiation of Pseudomonas aeruginosa strains by ribotyping: high discriminatory power by using a single restriction endonuclease[J]. J Med Microbiol,1994,40: 275 - 281.
[33]FerrusMA, HernandezM, HernandezHJ. Ribotypingof Pseudomonas aeruginosa from infected patients: evidence of common strain types[J].APMIS,1998,106(4):456-462.
[33]Grundman H,Schneider C,Hartung D,et al.Discriminatory power of three DNA-based typing technique of Pseudomonas aeruginosa[J].J Clin Microbiol,1995,33:528-534.
[34]Schwartz DC,Cantor CR.Separation of yeast chromosome-sized DNAs by pulsed-field gradient gel electrophoresis[J]. Cell,1984,37:67-75.
[35]RomlingU, TummlerB. Achieving 100% typeability of Pseudomonas aeruginosa by pulsed-field gel electrophoresis[J].J Clin Microbiol,2000,38:464465.
[36]Tenover FC, Arberit RD, Goering RV, et al. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis criteria for bacterial strain typing[J].J Clin Microbiol,1995,33(9):2233-2239.
[37]Speert D.Molecular epidemiology of Pseudomonas aeruginosa[J].Front Biosci,2002,7:354-361.
[38]Lewis DA,Jones A,Parkhill J,et al.Identification of DNA markers fora transmissible Pseudomon asaeruginosa cystic fibrosis strain[J].Am J Respir Cell Mol Biol,2005,33:56-56.
Overview of Molecular Typing Methods for Pseudomonas aeruginosa
XING Jin,YUE Bing-fei,HE Zheng-ming
(National Institutes for Food and Drug Control,Institute of Laboratory Animal Resources,Beijing 100050,China)
Pseudomonas aeruginosa is a common zoonotic opportunistic pathogen,widely distributed in nature,is one of important pathogens causing experimental animal pollution and intra-hospital infection.Molecular typing is an important tool in pathogen epidemiology.It is very effective for determining the source of infection and ways to detect crosscontamination and epidemic strains.In this paper,we summarized the research progress of P.aeruginosa molecular typing methods.
Pseudomonas aeruginosa;molecular typing methods;Laboratory animals
R33
A
1671-7856(2012)08-0062-06
10.3969.j.issn.1671.7856.2012.008.015
2012-05-28
邢进(1979-),男,助理研究员,研究方向:实验动物微生物检测。E-mail:xjvet@nifdc.org.cn。
贺争鸣,男,研究员,E-mail:zhengminghe@163.com。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
红外技术(2022年4期)2022-04-25
昆明医科大学学报(2022年2期)2022-03-29
中国医学装备(2020年4期)2020-05-23
中国中医急症(2019年10期)2019-05-21
中国医学装备(2019年1期)2019-02-14
中国新技术新产品(2018年22期)2018-01-05
中华骨与关节外科杂志(2017年1期)2017-05-17
中央民族大学学报(自然科学版)(2015年1期)2015-06-11
智能制造(2015年4期)2015-05-12
