
茴拉西坦及其制剂含量测定方法的改进
2012-05-07 02:27郭志渊徐燕傅萍
四川生理科学杂志 2012年2期
郭志渊 徐燕 傅萍
(四川省食品药品检验所,四川 成都 610097)
茴拉西坦为脑功能改善药,曾用名阿尼西坦,通过血脑屏障选择性作用于中枢神经系统。现行标准茴拉西坦及其制剂的含量测定均采用以甲醇-水为流动相的HPLC法或紫外分光光度法进行测定[1-4];文献报道茴拉西坦及其制剂的检测方法也以甲醇-水为流动相[5-7]。在检测中发现茴拉西坦样品在甲醇及流动相中的稳定性相当差影响到最后的检测结果。本文采用以乙腈-水(35:65)为流动相的HPLC法对茴拉西坦的含量进行测定,大大提高了样品溶液的稳定性,使测定结果更准确、可靠。
1 材料与方法
1.1 材料与仪器
Waters2695-2487高效液相色谱仪,Agilent1200高效液相色谱仪;茴拉西坦原料(晋城海斯制药有限公司提供);乙腈为色谱纯。
1.2 方法
1.2.1 色谱条件
色谱柱:Altima C18柱 250×4.6mm 5μm,Agilent EclipssXDB-C18柱250×4.6mm 5μm,流动相:乙腈-水(35:65);检测波长采用283nm,理论板数以茴拉西坦计算应不低于1500。
1.2.2 测定方法
取本品适量,精密称定,加流动相使溶解并稀释制成每1ml中含茴拉西坦0.08mg的溶液,作为供试品溶液。精密量取10μl注入液相色谱仪,记录色谱图;另取茴拉西坦对照品,同法处理作为对照品溶液。
1.2.3 专属性试验
吸取对照品溶液、供试品溶液、及按供试品溶液制备方法制成的空白辅料溶液各10μl注入液相色谱仪。照1.2.1方法测定。
1.2.4 线性关系
精密称取对茴拉西坦对照品19.48mg置25ml量瓶中,加流动相溶解并稀释至刻度,摇匀,精密量取1、2、5、10、20、40、80ml分别置100ml量瓶中用流动相稀释至刻度。精密量取10 μl,注入液相色谱仪,照1.2.1方法测定。
1.2.5 精密度试验
精密量取对照品溶液10μl,连续进样6次,照1.2.1方法测定。
1.2.6 重现性试验
取同一批6份,制备供试品溶液,照1.2.1方法测定。
1.2.7 稳定性试验
取本品供试品溶液分别在0、1、2、4、8h进样,照1.2.1方法测定。
同时照1.2.1方法,以甲醇-水(60:40)为流动相测定供试品溶液在0、1、2、4、8h的稳定性。
1.2.8 回收率试验
按处方比例精密称取一定量的空白辅料(对应于茴拉西坦10mg)9份,分别置10ml量瓶中,加流动相溶解并稀释至刻度,摇匀,精密量取2ml置25ml量瓶中,各精密加入茴拉西坦对照品溶液(浓度0.9212mg·ml-1)1.6ml、2.0ml、2.4ml,加流动相稀释至刻度,照1.2.1方法测定。
1.2.9 样品含量测定
取各剂型的样品照1.2.2测定方法测定。
2 结果
2.1 专属性试验结果
结果空白辅料溶液在与对照品色谱相应的位置上无吸收,证明空白辅料溶液无干扰,对照品、供试品及空白溶液色谱图见图1~3。
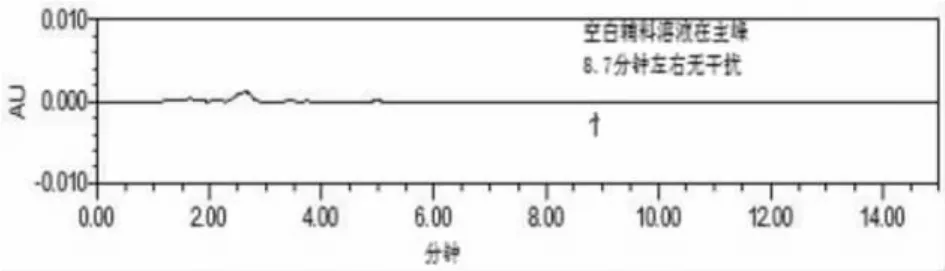
图1 空白辅料溶液色谱图

图2 供试品溶液色谱图
2.2 线性关系测定结果
以进样量(μg)为横坐标,峰面积为纵坐标进行线性回归,直线回归方程为 Y=2.7004×106X-4.6914×104,R=1.0000。由回归方程和直角坐标图可见,茴拉西坦进样量在0.07792~6.2336μg范围内,进样量与峰面积呈良好的线性关系。

图3 对照品溶液色谱图
2.3 精密度试验测定结果
以测得峰面积,计算RSD0.14%,精密度良好。
2.4 重复性试验测定结果
计算得茴拉西坦的平均含量为100.6%,RSD为0.97%。
2.5 稳定性试验测定结果
8小时内茴拉西坦峰面积的RSD为0.46%,表明供试品溶液在8小时内稳定,能满足测定需要。而当流动相为甲醇-水(60:40)时8小时内茴拉西坦峰面积的RSD为1.76%。

表1 乙腈-水(35∶65)为流动相的稳定性
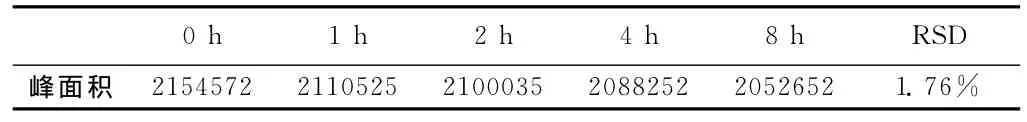
表2 甲醇-水(60∶40)为流动相的稳定性
回收率测定结果 通过峰面积,计算回收率,茴拉西坦的平均回收率为100.2%,RSD1.6%。
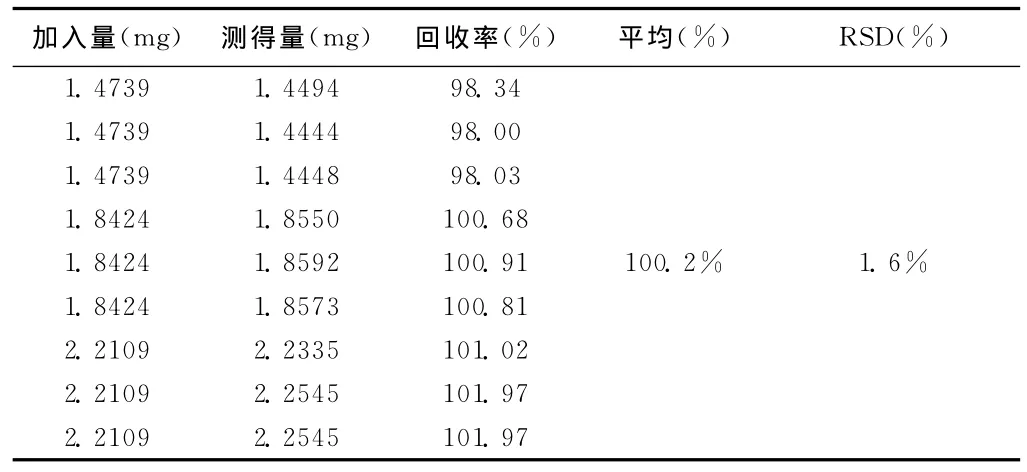
表3 回收率试验
2.7 样品测定结果
结果见表4。

表4 样品测定
3 讨论
3.1 流动相的选择
本实验考察了不同的流动相及相应的溶剂:甲醇-水(60:40)、乙腈-水(35:65)。发现当以甲醇-水(60:40)为流动相及溶剂时,茴拉西坦8小时内峰面积RSD为1.76%,样品稳定性差,不能满足测定需求;而用乙腈-水(35:65)为流动相及溶剂时,样品稳定性较好,8小时内峰面积RSD为0.46%,测定精密度大大提高。所以本文最终选择以乙腈-水(35:65)为流动相。
3.2 检测波长的选择
茴拉西坦及其制剂的原标准[1-4]及文献报道的茴拉西坦含量测定方法[5,7]的检测波长均不一致,并且考虑到改换溶剂后样品的最大吸收也会发生位移,所以本实验也对以乙腈-水(35:65)为溶剂的供试品溶液进行了紫外扫描,发现茴拉西坦在283nm处有最大吸收。故选283nm为检测波长。
综上所述,本文建立的茴拉西坦及茴拉西坦系列制剂的含量测定方法改变了现有标准[1-3]的流动相及检测波长,结果重现性更好,更准确可靠。
1 国家药典委员会.新药转正标准[S].第25册.北京:人民卫生出版社,2002,965-968.
2 国家药典委员会.新药转正标准[S].第46册.北京:人民卫生出版社,2002,21-21.
3 国家药典委员会.新药转正标准[S].第51册.北京:人民卫生出版社,2005,183-183.
4 国家药典委员会.新药转正标准[S].第65册.北京:人民卫生出版社,2009,134-134.
5 娄志红,苑淑俐.HPLC法测定茴拉西坦胶囊含量[J].黑龙江医药,2006,19(5):333-334.
6 许晋星.HPLC法测定茴拉西坦分散片的有关物质[J].广东药学院学报,2009,25(4):373-375.
7 刘东权,王晓文,陈鑫,等.HPLC法测定茴拉西坦咀嚼片中的含量[J].中国医药指南,2012,10(02):19-21.
猜你喜欢
煤化工(2022年3期)2022-07-08
昆明医科大学学报(2021年10期)2021-12-02
食品安全导刊(2021年21期)2021-08-30
科学与财富(2021年35期)2021-05-10
食品安全导刊(2020年14期)2020-12-04
中华养生保健(2020年1期)2020-11-16
食品安全导刊·中旬刊(2020年5期)2020-06-04
作文周刊·小学四年级版(2018年40期)2018-04-09
中国资源综合利用(2016年10期)2016-01-22
中国当代医药(2015年30期)2015-03-01
