
臭氧在氧化铝薄膜生长过程中氧化机理的研究
2012-01-05 08:53李淑梅
河北工业科技 2012年4期
李淑梅
(河北能源职业技术学院矿产资源与建筑工程系,河北唐山 063004)
臭氧在氧化铝薄膜生长过程中氧化机理的研究
李淑梅
(河北能源职业技术学院矿产资源与建筑工程系,河北唐山 063004)
使用密度泛函的B3LYP方法研究了臭氧在原子层沉积制备氧化铝过程中的初始氧化机理,计算发现:臭氧的端位O应该首先与Al原子配位成键,然后再与—CH3上邻近的H原子结合,最后释放O2,形成Al OH基团,因为这一路径是能量上更加有利的过程。
臭氧;反应路径;氧化铝;密度泛函理论
氧化铝薄膜具有很多优异的物理性能,如较高的介电常数、高热导率、高抗辐射性、高化学稳定性、对碱离子杂质低渗透性以及在很宽的波长范围内透明。因此,氧化铝薄膜广泛应用于微电子器件、电致发光器件以及抗腐蚀涂层等材料领域[1-2]。特别是,氧化铝薄膜由于高的介电常数(k≈9)被作为新型的高介电常数栅介质材料得到了广泛关注,因为相比氧化铪、氧化锆等高介电常数栅介质,氧化铝的突出优势是当沉积在硅衬底上时,不容易在衬底表面形成二氧化硅界面层。
利用原子层沉积制备氧化铝薄膜最早使用的是金属前驱物AlCl3,但由于会产生腐蚀性的副产物HCl,所以逐渐被三甲基化铝(TMA)取代。到目前为止,TMA已成为应用最广泛的金属前驱物。制备氧化铝薄膜应用的氧源主要有水、双氧水、臭氧以及一些金属氧化物。其中,臭氧由于它的高反应活性而得到了广泛应用。一直以来,臭氧的氧化机理在实验上都受到特别的重视。例如:PRECHTL等认为臭氧和—Al(CH3)2反应生成C2H4,并最终生成表面—OH,这样—OH可以进一步与TMA反应[3]。GOLDSTEIN等通过傅里叶转换红外光谱(FTIR)研究发现,表面AlCH3在与O3反应的过程中被消除,AlCH3被转化为 Al OCH3,Al(OCHO),Al(OCOOH)和 Al OH 等基团[4]。同时,四极质谱(QMS)分析显示臭氧反应主要产物是CH4,并伴随少量的C2H4,CO2和CO。ROSE等也通过QMS分析认为除了上述副产物外,H2O也是副产物之一[5]。虽然臭氧的作用机理在实验上已经被广泛研究,但是由于其反应机理复杂,所以很少有相关的理论计算方面的结果报道。笔者主要着眼于原子层沉积法制备Al2O3薄膜的臭氧脉冲阶段的初始氧化作用机理,通过建立臭氧与硅表面AlCH3的反应模型进行理论计算,计算结果将为进一步深入研究进行理论上的尝试。
1 计算细节和模型
全部计算由Gaussian 03程序包[6]完成,计算方法采用当今最流行的杂化密度泛函方法B3LYP,该方 法 结 合 了 BECKE 的 三 参 数 交 换 泛 函[7-8]和LEE等的相关泛函[9]。计算模型选取 Si9H12-O2AlCH3簇 (见图 1 )。在很 多 理 论 计算中[10-12],Si9H12单二聚簇通常被作为基本的Si模型来模拟Si(100)-2×1表面。这个单二聚簇的结构组成如下:4层Si原子,其中第1层2个表面Si原子,第2层4个Si原子,第3层2个Si原子,第4层1个Si原子。对其中不饱和的Si原子用H原子饱和,目的是阻止未配对电子在Si表面的转移,进而影响对Si表面的合理模拟。
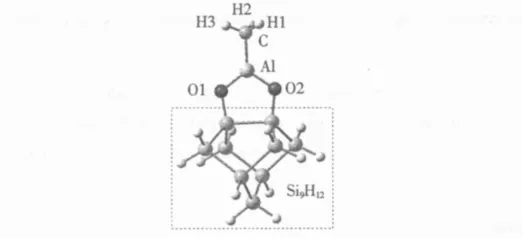
图1 Si9 H 12-O2 AlCH 3簇模型Fig.1 Si9 H 12-O2 AlCH 3 cluster model
在本研究中,使用混合基组方式以节省计算时间。第1层2个表面Si原子和—CH3,Al,O采用6-31+G(d)基函数,第2层、第3层及第4层Si原子以及用于饱和的H原子采用小的6-31G基组。几何结构采用全优化方法,没有作任何限制,在优化结构的基础上进行频率分析计算,来验证各个驻点在反应势能面上的性质。在局部极小点所有频率为正值,而过渡态[13]有一个虚频出现。计算得到的能量值都进行了零点能修正。
2 结果与讨论
图2显示了臭氧对Si9H12-O2AlCH3氧化的2个可能的反应路径以及反应物—过渡态—产物所对应的能量值。第1个反应路径(路径1)是O3的端位O直接从—CH3夺取1个 H原子,生成—AlCH2以及—OH和O2。第2个反应路径(路径2)是O3的端位O先与Al原子成键,然后再夺取—CH3的1个 H 原子,生成 Al(CH2)OH 和 O2。从图2可以看到,路径1的活化能是184.2 kJ/mol,路径2的活化能是127.6 kJ/mol。假设2个反应路径的指前因子相同,用阿累尼乌斯公式粗略计算2个反应路径的速率常数之比:

由计算的活化能可估计在300℃时,反应路径2的速率常数是反应路径1的1.4×105倍。因此,从动力学角度认为,反应路径2在能量上是更加有利的。
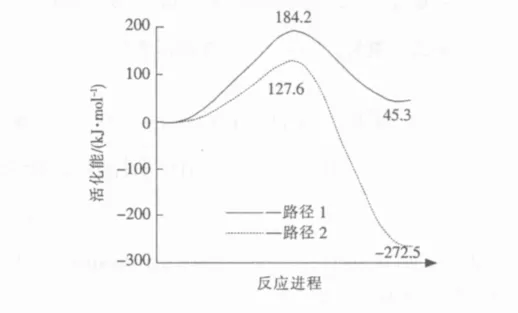
图2 臭氧对Si9 H 12-O2 AlCH 3的氧化反应路径及其能量变化Fig.2 Reaction pathways and the corresponding energy values of stationary points for the oxidation reactions of O3 and Si9 H 12-O2 AlCH 3
另外,从图2也能发现,反应路径1是一个吸热反应,共吸热45.3 kJ/mol,而反应路径2是一个强烈的放热反应,反应热是-272.5 kJ/mol。由此,可以得出结论,无论是从动力学还是热力学角度讲,反应路径2是最可能的路径。
通过反应路径上每一个驻点的振动分析,反应物和产物为势能面上的稳定结构,反应路径1和反应路径2过渡态计算得到的虚频分别是753i cm-1和1 383i cm-1。图3给出了2个反应路径上过渡态结构示意图,图中的箭头表示虚频的振动方向,从这里可以判断出过渡态可以很好地连接反应物和产物。
另外,从图3也可以定性地看到:反应路径1,最后生成的产物是AlCH2—,—OH和O2;而反应路径2中,最后生成的产物是AlCH2—,—AlOH以及O2。这一结果从表1也可以得到验证,通过和反应物比较,可发现过渡态的O3—O4距离增大很
3 结 论
从理论上,笔者给出了臭氧对AlCH3的2个氧化反应路径,这2个反应路径都是O3的端位O原子夺取—CH3的1个H,最后生成O2和—OH,但是又有本质的区别,一个形成的是自由—OH,一个是—Al OH。从反应的能量变化综合比较可以看出,O3的端位O应该首先与Al原子配位成键,然后再与—CH3上邻近的H原子结合,最后释放O2并形成—AlOH。因为这一路径是能量上更加有利多,趋向断开。于是,能够判断最后的产物有—OH和O2。当然这些生成的中间产物如何进一步反应是一个复杂的过程,将在笔者未来的研究中涉及。
反应路径1和路径2中反应中心相关键的键长见表1。从表1发现,反应物—过渡态—产物这一过程,C—Al和O4—O5基本上没有变化,所以不是反应的中心。O3—O4和C—H1逐渐增大,直到完全断开,而O3—H1逐渐减小,直至完全成键。这些键长的变化过程与反应的过程是一致的。的过程。

图3 臭氧对Si9 H 12-O2 AlCH 3的氧化反应路径的过渡态结构示意图Fig.3 Geometries of the transition state for the oxidation reaction paths of O3 and Si9 H 12-O2 AlCH 3
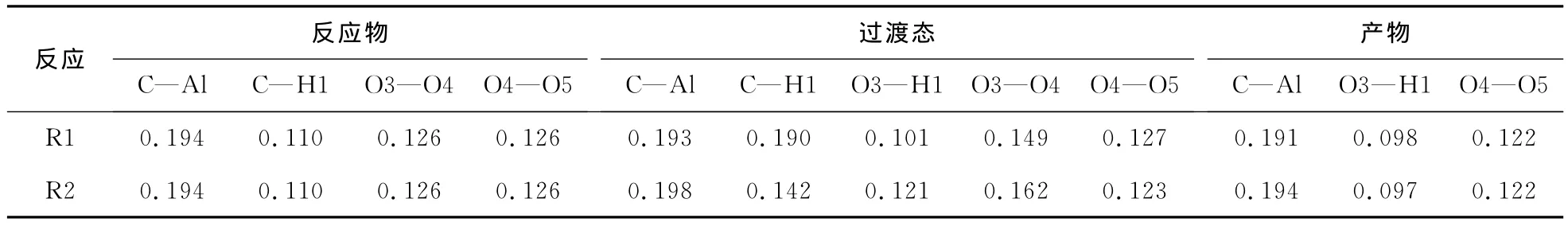
表1 反应路径1和反应路径2中反应中心相关键的键长Tab.1 Bond distances of the reactive center in the reaction pathways 1 and 2 nm
[1]卢红亮,徐 敏,丁士进,等.原子层淀积Al2O3薄膜的热稳定性研究[J].无机材料学报,2006,21(5):1 217-1 222.
[2]廖国进,巴德纯,闻立时,等.氧化铝薄膜在发光方面的研究进展[J].材料导报,2006,20(5):26-29.
[3]PRECHTL G,KERSCH A,ICKING-KONERT G S,et al.A model for Al2O3ALD conformity and deposition rate from oxygen precursor reactivity[A].IEEE International Electron Devices Meeting[C].[S.l.]:[s.n.],2003.961-964.
[4]GOLDSTEIN D N,MCCORMICK J A,GEORGE S M.Al2O3atomic layer deposition with trimethylaluminum and ozone studied by in situ transmission FTIR spectroscopy and quadrupole mass spectrometry[J].J Phys Chem C,2008,112:19 530-19 539.
[5]ROSE M,NIINISTÕJ,ENDLER I,et al.In situ reaction mechanism studies on ozone-based atomic layer deposition of Al2O3and Hf O2[J].ACS Appl Mater Interfaces,2010,2(2):347-350.
[6]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 03,Revision D.01[CP/CD]Wallingford:Gaussian Inc,2004.
[7]BECKE A D.Density-functional exchange-energy approxima-tion with correct asymptotic behavior[J].Phys Rev A,1988,38(6):3 098-3 100.
[8]BECKE A D.Density-functional thermochemistry(Ⅲ):The role of exact exchange[J].J Chem Phys,1993,98(7):5 648-5 652.
[9]LEE C,YANG W,PARR R G.Development of the colle-salvetti correlation-energy formula into a functional of the electron density[J].Phys Rev B,1988,37(2):785-789.
[10]REN J,SUN B,ZHANG D W.Density functional study of initial HfCl4adsorption and decomposition reactions on silicon surfaces with SiON interfacial layer[J].Appl Surf Sci,2007,253(23):9 148-9 153.
[11]REN J,ZHOU G F,HU Y Q,et al.Surface reaction mechanism of Y2O3atomic layer deposition on the hydroxylated Si(100)-2×1:A density functional theory study[J].Appl Surf Sci,2009,255(16):7 136-7 141.
[12]周广芬,崔永琴,于战海,等.HfO2和Al2O3混合原子层沉积反应机理的密度泛函理论研究[J].河北科技大学学报,2008,29(2):128-132.
[13]郭子成,任 杰,杨建一.等温、等容化学反应平衡时平衡关系的讨论[J].河北科技大学学报,2009,30(1):25-29.
Theoretical study on oxidation mechanism in depositing alumina thin films using ozone as oxygen source
LI Shu-mei
(Mineral Resources and Building Engineering Department,Hebei Energy College of Vocation and Technology,Tangshan Hebei 063004,China)
The initial oxidation mechanism of atomic-layer deposited alumina using ozone as oxygen source is studied by using density functional theory B3LYP method.Calculations show the following reaction pathway is energetically more favorable:the end oxygen atom of ozone coordinates the Al atom,then recombines with the neighboring hydrogen atom of CH3group,finally releases oxygen molecule to form AlOH group.
ozone;reaction pathway;alumina;density functional theory
O641
A
1008-1534(2012)04-203-03
2011-11-07;
2012-03-02
王海云
李淑梅(1971-),女,河北唐山人,高级讲师,硕士,主要从事物理化学及煤化工、有机化学方面的研究。
猜你喜欢
山东冶金(2022年4期)2022-09-14
北京航空航天大学学报(2022年5期)2022-06-06
建材发展导向(2021年14期)2021-08-23
氯碱工业(2020年9期)2020-03-02
中国煤层气(2019年2期)2019-08-27
电脑知识与技术(2018年3期)2018-03-21
环境保护与循环经济(2017年4期)2018-01-22
哈尔滨理工大学学报(2017年1期)2017-04-08
环境与可持续发展(2017年2期)2017-04-06
科技视界(2016年24期)2016-10-11
