
基于密度泛函理论解读不同高密度储氢材料释氢能力*
2011-10-23 12:13肖明珠张国英路广霞朱圣龙
物理学报 2011年2期
张 辉 肖明珠 张国英 路广霞 朱圣龙
1)(沈阳师范大学物理科学与技术学院,沈阳 110034)
2)(中国科学院金属研究所金属腐蚀与防护国家重点实验室,沈阳 110016)
(2009年10月22日收到;2010年5月13日收到修改稿)
基于密度泛函理论解读不同高密度储氢材料释氢能力*
张 辉1)†肖明珠1)张国英1)路广霞1)朱圣龙2)
1)(沈阳师范大学物理科学与技术学院,沈阳 110034)
2)(中国科学院金属研究所金属腐蚀与防护国家重点实验室,沈阳 110016)
(2009年10月22日收到;2010年5月13日收到修改稿)
采用基于密度泛函理论的第一原理平面波赝势方法,研究了MgH2,LiBH4,LiNH2,NaAlH4几种高密度储氢材料及其合金的释氢及影响机理.结果表明:高密储氢材料 MgH2,LiBH4,LiNH2,NaAlH4都比较稳定,释氢温度都很高,合金化可以降低它们的稳定性,但系统稳定性不是决定高密度储氢材料释氢性质的关键因素;带隙的宽窄基本可以表征储氢材料成键的强弱,能隙越宽,键断开越难,释氢温度就越高;LiNH2价带顶成键峰主要由Li—N成键贡献,N—H键构成较低的峰,使得LiNH2储氢材料的带隙虽很窄释氢温度却较高,且放氢过程中有氨气放出;合金化使得几种高密度储氢材料的带隙变窄,费米能级进入导带,从而使它们的释氢性能大大改善;电荷布居分析发现LiBH4中B—H键最强,LiNH2中H—N键最弱,因此LiNH2中H相对容易放出.合金化后,各储氢材料中X—H键强度都有所降低,且LiMgNH2中N—H键强度最低,因此从降低释氢温度角度,发展LiNH2储氢材料最为有利.
储氢材料,第一原理,释氢能力
PACS:61.66.Fn,71.17.Mb,71.20.Ps,65.40.-b
1.引 言
20世纪70年代以后,氢能的开发和利用进入了一个新的阶段,尤其是90年代后,一些国家加强氢能的开发和研究.据报道,美国能源部将氢能的研究经费中的50%用于储氢研究[1].氢的储存主要有三种方式:气态、固态和液态.理想的固体储氢材料应满足储氢密度高,释氢温度低,且储放氢速度快.国际能源组织提出的汽车燃料电池用储氢材料性能满足的目标是释氢温度低于100℃,可逆储氢量(质量分数)2010年达到 6.0%,2015年达9.0%[2].传统的MgH2和金属络合物储氢材料储氢密度较高(见表1),为使这些材料满足车载固体储氢材料要求,各国研究人员做了大量的工作,发现除储氢量外,它们都不能满足要求.从表1可以看出,MgH2虽具有储氢量高、质量小和成本低的特点,但其释氢温度高,其储放氢动力学性能也较差[3].通过纳米化和元素替代可以改善 MgH2的性能.NaAlH4在加入催化剂时能在低于100℃下可逆吸/放大量氢气,氢气的纯度高,可循环使用,催化剂价格便宜,但它的实际储放氢量不足5%[4].Chen等[5]提出氮化锂可以大量可逆地吸放氢,从而引发了众多学者对 Li—N—H系作储氢材料的研究.氮化锂的储氢反应方程为
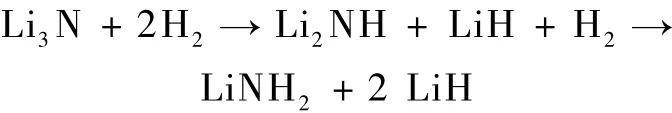
该反应总的储氢量为10.4%,后一步反应储氢为6.5% ,在 200—320 ℃ 放氢量是 6.3%[5].可见 Li—N—H储氢材料具有较高的储氢量和较温和的释氢条件,但放氢温度还是较高.通过加入催化剂和元素替代可以改善 Li—N—H储氢材料的释氢性能[6,7].LiBH4的储氢量高达 18.3% ,可以满足下一代燃料电池所需的目标,但它相当稳定,300—600℃释氢只有8%.可见几种高密度储氢材料的储放氢性能都不能满足车载燃料电池的需要,尚需要继续做大量研究工作.目前的研究工作有些是从实验的角度靠经验摸索,太耗费人力物力.亟待加强理论研究,一种实验现象出现有一理论解释才完美,同时理论研究也可指导实验研究.不同储氢材料具有不同储氢性能,有其内在的根据,他们的储放氢性能改善也有规律可循.本文将利用第一性原理的方法对 MgH2,NaAlH4,LiNH2,LiBH4四种高密度储氢材料进行研究,以期从电子尺度解读它们的释氢机理和影响释氢的机理,以此对不同储氢材料进行筛选并指导如何改善储氢材料的性能.

表1 四种储氢材料理论储氢量(质量分数)和释氢温度
2.计算模型和理论方法
2.1.晶体结构
MgH2的晶体结构如图1(a)所示,晶格常数为a=b=0.4501 nm,c=0.3010 nm,空间群为 P42/mnm;LiBH4晶体空间群为 Pnma,晶格常数 a=7.1786 nm,b=4.4369 nm,c=6.8303 nm(见图 1(b));LiNH2的晶体结构如图1(c)所示,晶格常数为a=b=5.037 nm,c=10.278 nm,空间群为 I-4;NaAlH4属I-41/a空间群,晶格常数为 a=b=5.044 nm,c=11.434 nm,(见图 1(d)).
2.2.理论方法
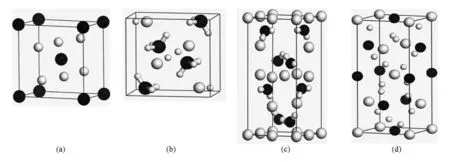
图1 四种高密度储氢材料的原子结构模型 (a)MgH2,黑色球为Mg,白色球为H,(b)LiBH4,大白球为Li,小白球为H,大黑球为 B;(c)LiNH2,小白球为 H,黑色球为 B,大白球为 Li;(d)NaAlH4,小白球为 H,大白球为Al,黑色球为 Na
采用基于密度泛函理论的“总体能量-赝势平面波方法”(Castep软件包)[8],交换关联能采用广义梯度近似(GGA),采用对正则条件进行弛豫的超软赝势[9]作为平面波基集.采用自洽迭代(SCF)方法进行计算时,采用结合BFGS共轭梯度方法的Pulay密度混合方案[10]处理电子弛豫.先对模型的晶体结构进行完全的几何优化,以求得它们的局域最稳定结构.优化结束时,体系总能量的收敛值取1.0×10-5eV,作用每个原子上的力低于0.3 eV/nm,公差偏移小于1.0×10-4nm,应力偏差小于0.05 Gpa.
3.结果分析与讨论
3.1.结合能
晶体的结合能是指晶体分解成孤立原子时需要的能量,可用来表征晶体结构的稳定性.表2给出了四种储氢材料及其合金的结合能,MgH2,LiBH4,LiNH2,NaAlH4结合能都较大,说明它们都很稳定,因此需较高的释氢温度.通过掺杂发现络合物储氢合金(MgH2除外)的结合能都有所降低,可见合金化可以改善储氢材料的释氢性能.
从表2可知 LiNH2的结合能最大,为每原子4.779 eV,说明该材料最稳定,按此推理它的释氢温度应最高,但实际上LiBH4的释氢温度最高,可见从结合能不能解释材料的放氢机理.储氢材料放氢反应时,晶体不完全分解,主要是与氢结合的键断开.结合能不能反映各原子间结合的强弱.LiBH4的释氢温度高于 LiNH2,可能是由于 LiBH4中 B—H键强度比LiNH2中N—N键强度大,这一点我们在后面用态密度、电荷布居来讨论.
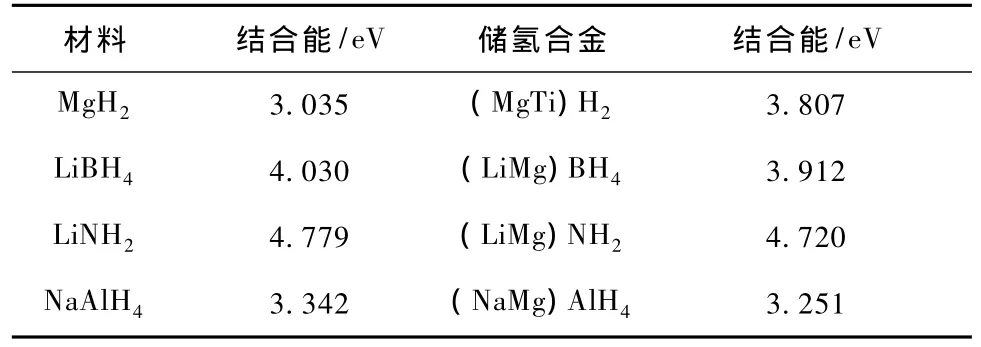
表2 四种储氢材料及其合金的结合能
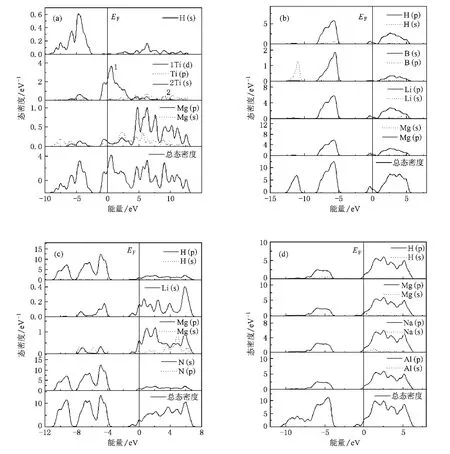
3.2.态密度
态密度对于分析材料中的原子成键和材料特性有重要的意义.计算给出了 MgH2,LiBH4,LiNH2,NaAlH4(见图2)及它们的合金(见图3)的总态密度及相应原子的分波态密度.从图2可以看出,几种储氢材料的能带都由价带、导带和带隙组成,且带隙较宽,费米能级位于价带顶,具有明显的绝缘体特征.带隙从宽到窄的顺序为 LiBH4,NaAlH4,LiNH2,MgH2,能隙越宽,说明此晶体中结合最弱的键上的电子被束缚得越强烈,这样这个键断开也就越难,因此可以用它表征晶体或团簇成键的强弱.由此可以得出LiBH4中结合最弱的键较其他储氢材料的强,所以其释氢温度最高.
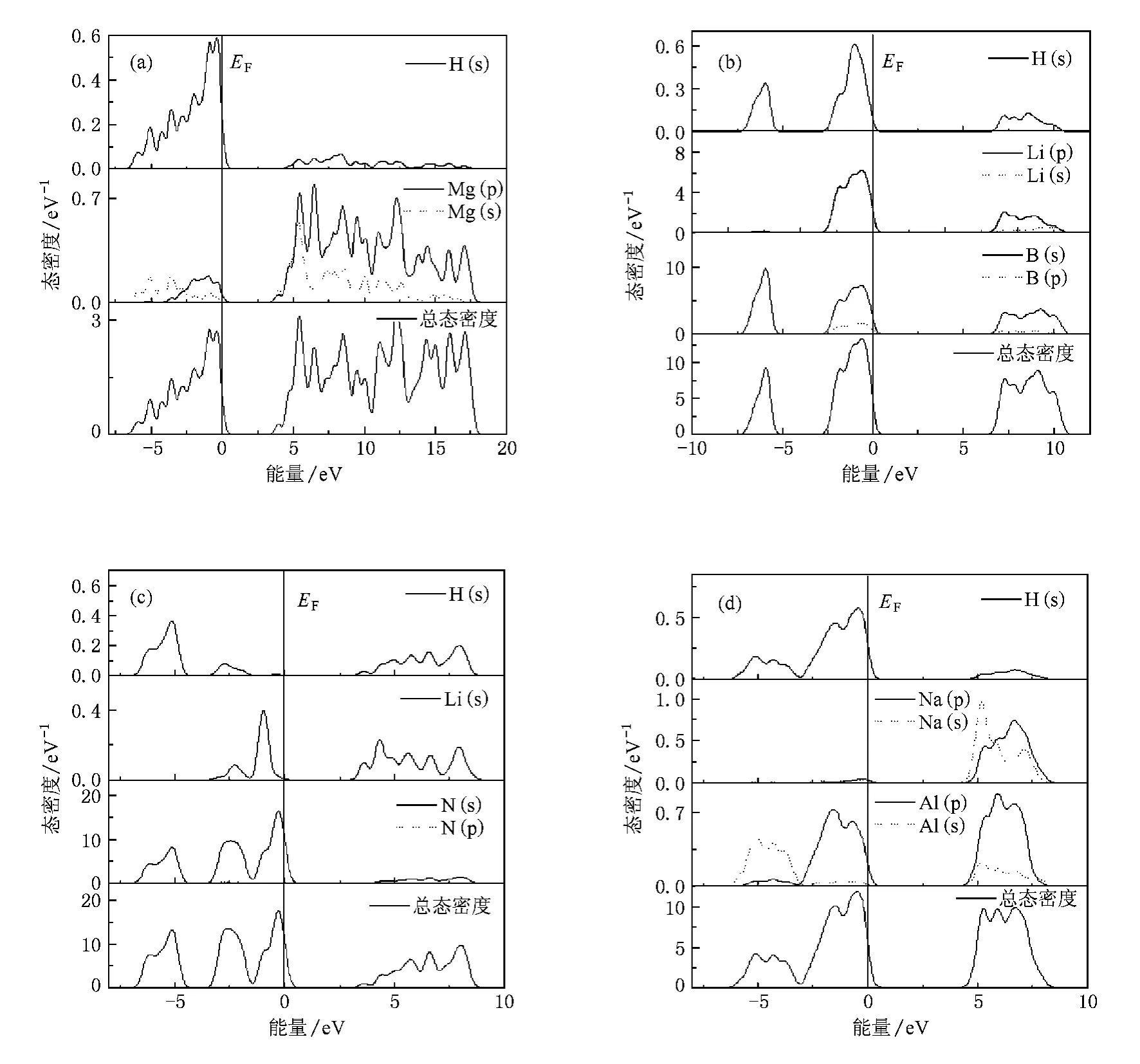
下面我们再看一下不同储氢材料中结合较弱的键(接近费米能级的成键峰)是由哪些原子形成的.储氢材料的释氢能力是由与氢结合的键决定的.图2(a)给出了MgH2的总态密度及各原子的分波态密度.可看出MgH2费米能级以下的成键峰是H(s),Mg(p)及少量 Mg(s)杂化而成的.LiBH4(见图2(b))价带有两个成键峰,低能区的由B—H成键贡献,接近费米能级的成键峰是B—H和B—Li成键而成.从LiNH2的总态密度和各原子的分波态密度可以看出(见图2(c)),LiNH2有三个成键峰,接近费米能级的成键峰主要是由Li—N成键贡献,而第二、第三个峰由N—H键贡献,N—H键断开需电子从费米能级下第二峰跃迁到导带,其能量间隔很大,因此LiNH2带隙虽很窄,其释氢温度也很高.在LiNH2中Li—N键强度弱,Li—N键断开,会留下N—H团簇,这可以解释 LiNH2释氢过程中总会伴随NH3的产生.图2(d)是NaAlH4的总态密度和各原子的分波态密度.NaAlH4的态密度分布与LiBH4类似,接近费米能级的成键峰主要是Al—H成键而成,只是其带隙比LiBH4的窄.
图 3 给 出 了 (MgTi)H2,(LiMg)BH4,(LiMg)NH2,(NaMg)AlH4合金的态密度图.4个图都有两个明显的特征,即费米能级都进入导带,带隙明显变窄.按上述观点:能隙越宽,晶体中结合最弱的键上的电子被束缚得越强烈,那么合金元素掺杂,储氢合金中结合最弱的键变得更弱,这样就降低了释氢温度.仔细观察图3中各图,发现MgH2掺Ti(见图3(a)),使原带隙中出现一尖峰,这使得MgH2的带隙大大降低,使得 H—Mg或 H—Ti键上电子(原价带顶电子)跃迁到导带变得非常容易,即H—Mg或H—Ti键容易断开,降低了释氢温度;再看掺 Mg的 LiBH4态密度图(见图3(b)),受掺杂Mg的影响,态密度原带隙接近导带底处出现一小尖峰,这同样使H—B或Li(Mg)—B键键强减弱,降低释氢温度;同样镁掺杂使 (LiMg)NH2(图3(c))和(NaMg)AlH4(图3(d))的带隙有所减小(对NaAlH4带隙影响大),从而使它们的释氢性质有所改善.
3.3.电荷布居
电荷布居可以对原子间成键的强弱进行定量说明,根据电荷布居的数值来判定原子之间成键的强弱.表3给出了四种材料及其合金的 X—H原子间的电荷布居数.从表3可以看出MgH2及其合金电荷布居数为负值,说明Mg—H,Ti—H键不是共价键.我们已经知道MgH2是离子化合物,所以我们的计算合理.其他几种储氢材料及其合金都是共价化合物.LiBH4中B—H的电荷布居为1.017,电荷密度重叠最大;LiNH2中H—N的电荷布居为0.76,电荷密度重叠最小;说明B—H表现为强共价键,H—N的共价键最弱,说明 LiBH4中氢最难放出,而LiNH2中H相对容易放出,这与实验结果是一致的.合金元素掺杂后,储氢材料中X—H键的电荷布居都有所降低,但LiMgBH4和NaMgAlH4的仍很高,因此从降低释氢温度角度,发展 LiNH2储氢材料最有利.
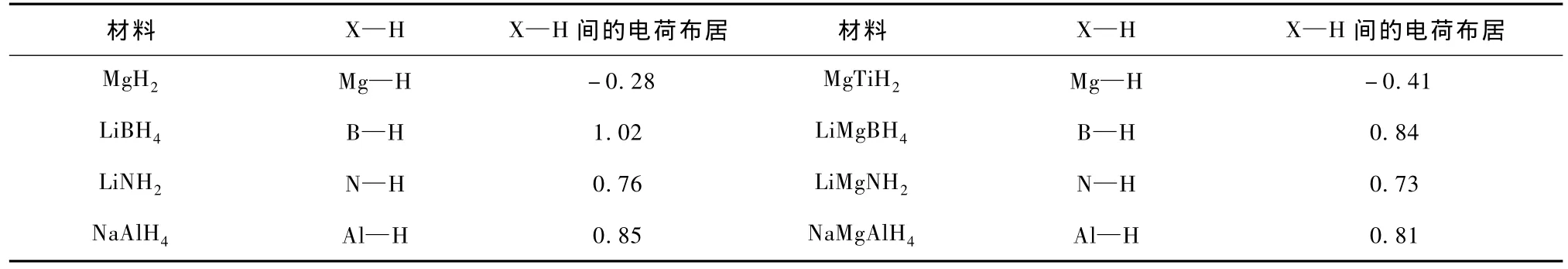
表3 四种材料及其合金中原子间的电荷布居
4.结 论
采用基于密度泛函理论的第一原理平面波赝势方法,研究了 MgH2,LiBH4,LiNH2,NaAlH4几种高密储氢材料及其合金的释氢及影响机理,得出以下结论.
1)MgH2,LiBH4,LiNH2,NaAlH4几种高密储氢材料都比较稳定,释氢温度都很高;通过合金化可以降低储氢材料的稳定性,改善其释氢性能.研究还发现影响高密度储氢材料释氢温度的关键因素不是系统稳定性.
2)通过对态密度分析发现带隙的宽窄基本可以表征储氢材料成键的强弱,能隙越宽,晶体中结合最弱的键上的电子被束缚得越强烈,断开也就越难.LiBH4带隙最宽,因此它的释氢温度也最高.LiNH2价带顶成键峰主要由 Li—N成键贡献,N—H键构成较低的峰,使得 LiNH2储氢材料的带隙虽很窄但释氢温度较高,且放氢过程中有氨气放出.
3)合金化使得几种高密度储氢材料的带隙变窄,费米能级进入导带,从而使它们的释氢性能大大改善.
4)电荷布居分析发现,MgH2是离子化合物,LiBH4,LiNH2,NaAlH4是共价化合物.LiBH4中 B—H键最强,LiNH2中H—N键最弱,因此LiNH2中 H相对容易放出.合金化后,各储氢材料中X—H键强度都有所降低,且 LiMgNH2中 N—H键强度最低,因此从降低释氢温度角度,发展LiNH2储氢材料最为有利.
[1]Bao D Y 1994J.Power Sour.16 1(in Chinese)[鲍德佑 1994新能源 16 1]
[2]Fang S S,Dong Y D 2001Chinese Journal of Nature23 259(in Chinese)[方守狮、董远达 2001自然杂志 23 259]
[3]Yao X D,Lu G Q 2008Chin.Sci.Bull.53 2421
[4]Zhuang P H,Liu X P,Li Z N,Wang S M,Jiang L J,Li H L 2007Trans.Nonferrous Met.Soc.China17 985
[5]Chen P,Xiong Z T,Luo J Z,Lin J Y,Tan K L 2002Nature420 302
[6]Zhang H,Qi K Z,Zhang G Y,Wu D,Zhu S L 2009Acta Phys.Sin.58 8077(in Chinese)[张 辉、戚克振、张国英、吴 迪、朱圣龙 物理学报2009 58 8077]
[7]Zhang H,Liu G L,Qi K Z,Zhang G Y,Xiao M Z,Zhu S L 2010Chin.Phys.B 19 048601
[8]Segall M D,Lindan P J D,Probert M J,Pickard C J,Hasnip P J,Clark S J,Payne M C 2002Phys.Condens.Matter14 2717
[9]Vanderbilt D 1990Phys.Rev.B 41 7892
[10]Hammer B,Hansen L B,Norkov J K 1999Phys.Rev.B 59 7413
PACS:61.66.Fn,71.17.Mb,71.20.Ps,65.40.-b
Interpretation of dehydrogenation ability of high-density hydrogen storage materials by density functional theory*
Zhang Hui1)†Xiao Ming-Zhu1)Zhang Guo-Ying1)Lu Guang-Xia1)Zhu Sheng-Long2)
1)(College of Physics Science and Technology,Shenyang Normal University,Shenyang 110034,China)
2)(State Key Laboratory for Corrosion and Protection,Institute of Metal Research,Chinese Academy of Sciences,Shenyang 110016,China)
(Received 22 October 2009;revised manuscript received 13 May 2010)
A first-principles plane-wave pseudopotential method based on the density functional theory was used to investigate the dehydrogenation properties and its influence mechanics on several high-density hydrogen storage materials(MgH2,LiBH4,LiNH2and NaAlH4)and their alloys.The results show that MgH2,LiBH4,LiNH2and NaAlH4high-density hydrogen storage materials are relatively stable and have high dehydrogenation temperature.Alloying can reduce their stability,but the stability of a system is not a key factor to the dehydrogenation properties of high-density hydrogen storage materials.The width of band gap of hydrogen storage materials can characterize the bond strength basically,the wider the energy gap is,the harder the bond breaks,and the higher the dehydrogenation temperature is.The bonding peak of the valence band top of LiNH2is attributed mainly to the Li—N bonding,the N—H bond constitutes the low peak,which makes the dehydrogenation temperature of LiNH2high,though LiNH2has a narrow band gap in respect to LiBH4and NaAlH4,which makes the ammonia release in the dehydrogenation process.Alloying makes the band gap narrow,and the Fermi level goes into the conduction band,which improves the dehydrogenation properties.It was found from the charge population analysis that B—H bond in LiBH4is the strongest,H—N bond in LiNH2is the weakest,so LiNH2is relatively easy to release hydrogen.After alloying,the bond strength of X—H is weakened in every hydrogen storage material,and the N—H bond strength in LiMgNH2is the lowest.Therefore,it is perspective to develop LiNH2as hydrogen storage from the lowering of dehydrogenation temperature.
hydrogen storage material,first-principles calculation,dehydrogenation ability
*国家高技术研究发展计划(批准号:2009AA05Z105)、辽宁省教育厅科学研究计划(批准号:2008S215,2009S099)和辽宁省自然科学基金(批准号:20102173)资助的课题.
*Project supported by the National High Technology Research and Development of China(Grant No.2009AA05Z105),the Science Research Program of the Education Bureau of Liaoning Province of China(Grant Nos.2008S215,2009S099)and the Natural Science Foundation of Liaoning Province,China(Grant No.20102173).
猜你喜欢
辽宁科技大学学报(2022年5期)2023-01-04
山西大学学报(自然科学版)(2022年5期)2022-11-23
中外文摘(2021年7期)2021-04-23
发明与创新(2020年39期)2020-10-15
原子与分子物理学报(2020年5期)2020-03-17
数学物理学报(2019年6期)2020-01-13
发明与创新·小学生(2019年12期)2019-12-05
成都信息工程大学学报(2019年3期)2019-09-25
科技创新与应用(2018年21期)2018-09-14
振动工程学报(2017年4期)2018-05-31
