
漆酶空间结构、反应机理及应用
2011-09-29 07:26:20葛宏华武赟肖亚中
生物工程学报 2011年2期
葛宏华,武赟,肖亚中
安徽大学现代实验技术中心&生命科学学院,合肥 230039
漆酶空间结构、反应机理及应用
葛宏华,武赟,肖亚中
安徽大学现代实验技术中心&生命科学学院,合肥 230039
漆酶 (EC 1.10.3.2) 属于多铜氧化酶家族,可以催化氧化酚类和芳香类化合物,利用自由基反应机理完成4个电子的转移,并将分子氧还原成水。漆酶具有非常保守的拓扑学结构,结合作者自身工作实践,对漆酶结构与功能的最新研究进展进行综述,其中对漆酶的三维结构、活性中心、催化机理研究和最新的应用进展作重点阐述。
多铜氧化酶,漆酶结构,三核中心
Abstract:Laccases (benzenediol: oxygen oxidoreductases; EC 1.10.3.2) are copper-containing polyphenol oxidases that can oxidize a wide range of aromatic compounds, concomitantly with the transfer of four electrons and the reduction of molecular oxygen to water. The progress on the research of laccases structure and function is reviewed. Their three-dimensional structures and catalytic mechanism, as well as their applications in different fields are emphasized.
Keywords:multicopper oxidases, laccase structure, trinuclear cluster
漆酶 (EC 1.10.3.2) 是一种含铜的多酚氧化酶,是蓝色多铜氧化酶家族 (MCO) 中最主要的成员。它利用铜离子特有的氧化还原能力氧化酚类和芳香类化合物,同时将分子氧还原成水[1]。在 200多种氧化还原酶当中,只有 6类酶可以催化这种有氧的反应 (细胞色素氧化酶、漆酶、L-抗坏血酸氧化酶、血浆铜蓝蛋白、胆红素氧化酶和吩恶嗪合成酶)。
漆酶最初发现于日本漆树的汁液中,之后,在各种担子菌与子囊菌中均发现有大量的漆酶存在,真菌漆酶具备多种生理生化功能,如形态发生、真菌与植物寄主相互作用、应激防御以及木质素降解[2]。目前,几乎所有的白腐真菌中均发现有漆酶的存在,同时该酶也广泛存在于植物、部分细菌和昆虫中。植物漆酶与木质素的生物合成密切相关;细菌漆酶主要参与形态发生、芽胞外被抵御紫外与过氧化物的伤害、孢子色素的生物合成和铜离子的体内平衡;而昆虫漆酶蛋白的主要功能是控制表皮的骨化作用[3-4]。
1 漆酶的结构与功能
1.1 一级序列与空间结构
大部分漆酶以胞外单分子球状蛋白形式存在,其分子量约为60~70 kDa,等电点pI 4.0左右。漆酶分子均有不同程度的糖基化,其含量为10%~25%,也有一些大于30%[5]。每个漆酶分子内含有4个铜离子,根据光谱学特性可以分成 3类,包括 1个 I型 (T1) 铜离子 (Cu1)、1个 II型 (T2) 和 2个 III型 (T3) 铜离子 (Cu2和Cu3),其中Cu2和Cu3形成了1个三核中心 (TNC)[6-7]。通过对100多个漆酶的序列比对和分析,发现有 4个特征性序列区(L1−L4) 可以用来鉴别漆酶[8](表1),12个与铜离子结合的氨基酸残基均位于这4个保守区域,其中L2和 L4与之前报道的 MCO含铜蛋白特征性序列相符,而L1和L3则是漆酶特有的序列区。此外,根据目前漆酶已知空间结构的比对,可以鉴定出与底物结合相关的 4个loop区 (loop I、II、III、IV) (表2)。相对于特征性序列区,这些loop区无论从一级序列还是空间位置及长度都有着较为明显的不同,导致了各种漆酶拥有不同的底物特异性。例如笔者解析的栓菌Trametes sp. AH28-2的LacB晶体结构(PDB注册号为3KW7) 具有较长的loop IV,大大延伸了底物结合区域,同时该loop带有较强的负电荷,并在漆酶分子表面形成了一个凸起,因此可能对调节底物的结合发挥着重要作用[9]。
迄今有下列担子菌漆酶的三维结构被解析:灰盖鬼伞Coprinus cinereus的漆酶Lac-Cc (CcL: pdb注册号 1A65)[10],变色栓菌 Trametes versicolor 的Lcc I (TvL: 1GYC)[11]和 Lac IIIb (TvL: 1KYA)[12],木硬孔菌 Rigidoporus lignosus的RlL (R1G: 1V10)[7],虎皮香菇Lentinus tigrinus的LtL (2QT6)[13],毛栓菌Trametes trogii的TtL (2HRG)[14],毛栓菌Trametes hirsuta的ThL (3FPX)[15]以及Trametes sp. AH28-2的LacB (3KW7)[9]。另外,有晶体结构报道的还有来自子囊菌热白丝菌 Melanocarpus albomyces的天然漆酶 MaL (1GWO) 及其重组形式蛋白 rMaL(2Q9O)[16-17]、以及个别细菌漆酶[4]。

表1 漆酶特异性序列Table 1 laccase signature sequences

表2 漆酶的底物结合loop序列比对Table 2 Structural alignment of substrate binding loops
所有的这些漆酶结构均采用一种相似的拓扑学结构,以笔者解析的Trametes sp. AH28-2的LacB晶体结构为例 (图 1) (本文中蛋白结构示意图均使用 Pymol软件绘制[18]),整个单体分子由 3个杯状(Cupredoxin-like) 结构域 (Domain 1、2、3) 组成,每个结构域均有1个希腊花边 (Greek key) β桶状结构。Cu1位于domain 3,而TNC则深埋于domain 1和3之间,并与它们的一些保守残基相互作用。此外,LacB分子内还有 2个二硫键连接着 domain 1和2以及domain 1和3,但也有些真菌漆酶含有3个二硫键,比如MaL[16]。和大部分漆酶一样,LacB是一个糖蛋白,糖的含量约25%,在预测的11个糖基化位点中,晶体结构清楚地显示有 8个位点被不同程度地糖基化[9]。

图1 Trametes sp. AH28-2的LacB晶体结构立体视图Fig. 1 Stereo view of Trametes sp. AH28-2 LacB structure.
1.2 Cu1结合位点及与还原性底物的反应
典型的MCO成员一般都有4个氨基酸残基 (通常有1个Met) 围绕着Cu1形成了第1个配位球状体结构[1],但在漆酶结构中,Cu1与 1个 Cys的硫原子、2个His的氮原子 (Nd1) 组成了1个平面三角配位体结构。Cys的硫和Cu的配位键在614 nm处有特征性吸收峰,这也是漆酶呈现蓝色的原因[19]。在漆酶催化反应中,底物的4个单电子被转移,而Cu1是初级电子受体位点,之后电子通过高度保守的His-Cys-His转移到TNC三核中心,进而传递给分子氧使之还原成水[20](图2A)。Cu1的还原是催化过程的限速反应,采取的是Marcus的“outer-sphere”机理,即Cu1与底物之间氧化还原电势差ΔE0决定了电子转移速率[21]。Cu1的E0基本上在420~790 mV之间,高于其他的多铜氧化酶[22],目前普遍认为漆酶Cu1位点缺乏第4个配位残基是造成这种高氧化还原电势的重要因素。但这些具有相似的Cu1配位体结构的漆酶成员如何呈现出不同的氧化还原电势还需要进一步阐明。
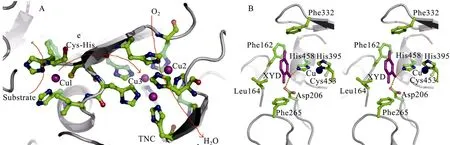
图2 漆酶活性中心结构示意图 (A) 及TvL1KYA与2,5-二甲苯胺相互作用立体示意图 (B)Fig. 2 The structure of the laccase active site (A) and stereo view of the active site of TvL1KYA, which binds substrate 2,5-dimethylaniline (B).
Cu1位点是底物反应场,位于漆酶的表面并形成了一个比较大的凹槽,从而能够宽松地容纳多种底物。结构TvL1KYA (TvL表明漆酶的来源,1KYA表示PDB注册号,下同) 第一次展示了高分辨率的漆酶与底物2,5-二甲苯胺形成的复合物结构[12],结构表明 2,5-二甲苯胺结合于底物结合区域,并于 4个loop上的若干疏水性残基发生相互作用 (图2B)。此外,His458和 Asp206在酶与底物的氨基相互作用中发挥重要作用,His458与Cu1配位成键并在所有MCO中高度保守,其侧链咪唑环与2,5-二甲苯胺上的氮形成氢键,这表明在电子传递给Cu1过程中,His458起到了一个入口的作用。与此同时,2,5-二甲苯胺上的氨基与结合区域背面的 Asp206的侧链氧形成了另一个氢键[16],在担子菌的真菌漆酶中,Asp206位点相当保守,而来自于子囊菌的漆酶在这个位置则用Glu替代了Asp。Asp与底物的相互作用决定了漆酶的pH依赖性[23]:当pH从3上升到5,由于Asp206侧链的去质子化,使得酚类化合物底物反应的Km值下降。在pH 5时,Asp206的羧基发生解离,使得活性位点带上负电荷,从而有利于带羟基或氨基的底物的结合[12]。
Piontek等认为Cu1与配位基团之间的距离与其氧化还原电势密切相关:较长的距离会产生较高的氧化还原电势[11]。在结构TvL1GYC中,由于Cu-N键的延长使得自由电子对铜的贡献降低,造成铜表现出更多的“缺电子”特性,引起这种较高氧化态的不稳定,因而提高了氧化还原电势。而Cu-N键的增加可能是由于 Ser113和 Glu460形成了氢键,导致His458远离了Cu1 (图3)。R1G1V10的结构也验证了上述猜想[7],该酶也同样属于高 E0的漆酶,其Glu459与 Ser113形成了一个很强的氢键,使得Cu1-His配位键的距离达到了 2.2Å。序列数据显示Glu460和Ser113在所有高电势漆酶中高度保守。根据TtL2HRG结构,Matera等[14]提出,在Cu1附近的2个疏水性氨基酸残基Phe460和Ile452导致了高氧化还原电势的产生,此外,Phe460周边大量的疏水性残基也可能对Cu1的高电势作出了贡献。
综上所述,漆酶Cu1的氧化还原电势不能归结于某单一结构,而是多种因素共同作用的结果,如Cu1的配位体结构和周边残基的特性。此外,底物的位阻效应也是影响漆酶反应偏好性的重要因素[24],来自T. villosa和M. thermophila的2个漆酶具有显著不同的氧化还原电势 (分别为0.79V和0.46V),2个苯丙氨酸 (Phe332和Phe265) 形成了进入活性位点的入口,因为它们之间距离的不同从而决定了进入活性位点的底物具有不同的氧化还原电势(图 2B)。

图3 TvL1GYC结构中由于Ser113和Glu460形成了氢键,导致His458远离了Cu1Fig. 3 The His458 moved a way from Cu1 as a result of the H bond between Ser113 and Glu460 in TvL1GYC structure.
1.3 TNC及其与分子氧的反应
另外3个铜离子分别属于Cu2和Cu3,以三角形式形成三核中心 (TNC),与 4个非常保守的His-X-His序列配位,其中6个组氨酸与2个Cu3形成配位键,另2个组氨酸与Cu2配位。Cu2离子呈顺磁性,而2个Cu3离子为耦合的离子对,为抗磁性。Cu3在330nm处有宽的吸收峰,Cu2则没有特征的吸收峰[25]。TNC的作用是结合分子氧,利用从单核中心Cu1传递过来的4个电子将分子氧还原成水[10]。对具体催化细节目前还比较有争议。Lee等认为[19]:从全还原状态的酶到氧分子的2个连续的双电子转移过程中产生 2个中间体形式,即过氧中间体 (PI:peroxy intermediate) 和天然中间体 (NI:native intermediate) (图4)。第一步双电子转移是限速步骤,而包括2个电子还原性的切割O-O键的第二步反应则快得多。处于完全还原状态时,TNC具有配体不饱和性[26],附近残基的阴离子电荷则稳定了带有高正电荷的TNC,特别是在Cu2附近高度保守的阴离子残基 (如在TvL1KYA的D77) 在这个反应过程中起着非常关键的作用[20,26]。NI时氧分子被完全还原成水但还保持在三核中心,其所有连接键的结构更有利于电子在三核中心重排[20]。相反,当酶处于静止态形式 (Resting form) 时,Cu2就会从这些连接键中脱离出来。在没有还原性底物存在时,NI衰减成静止态会非常缓慢,因此 NI应该是酶完全氧化时的形式[19]。事实上NI衰减成静止态过程涉及到氧原子从三核中心由内而外的结构重组,1个剩余的氧原子以OH−的形式与 Cu2结合,2个 Cu3核心也与 1个 OH−配位成键[27]。大致上,4个铜原子在还原底物的作用下,首先Cu1被还原,然后通过Cu-S(Cys)-N(His) 途径,将TNC的3个铜逐个还原。对于还原顺序,Yoon等提出了一个比较合理的机制[27],因为在Cu2和Cu3b附近存在有带负电荷的残基,因此Cu3a第一个被还原,之后,Cu2会被还原,剩下的Cu3b通过位于Cu1和Cu3核心之间的 Cys-His途径得以快速还原并伴随着 2个水分子的释放。如果Cu3b的还原早于Cu2就会导致OH−配位键的质子化,造成Cu2-Cu3b间电子耦合力的下降,最终无法将电子传递给Cu2。
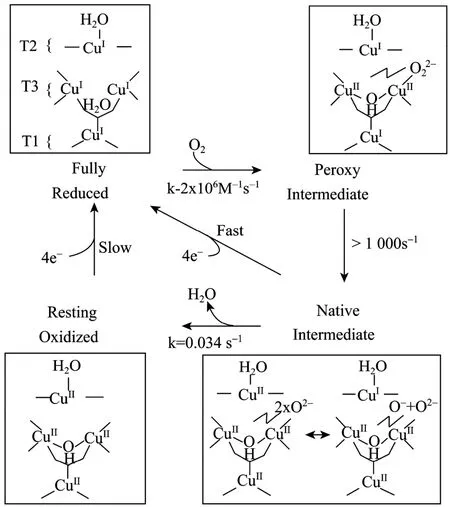
图4 MCO还原分子氧成水分子的机理Fig. 4 Mechanism of O2reduction to water by the MCOs.
在蛋白结构内部有2个通向TNC的溶剂通道,第1个指向2个Cu3离子,使得氧分子可以进入并结合TNC,第2个通道位于TNC另一边通向Cu2,当氧分子还原成水分子后从该通道释放[11,13]。
2 漆酶的反应及其应用前景
酚类化合物是典型的漆酶底物,它们的氧化还原电势较低,可以允许电子传递给 Cu1。丁香醛连氮 (Syringaldazine)、DMP和邻甲氧基苯酚是最常见的酚类底物,但漆酶也能氧化其他的一些电子供体底物如ABTS或氰亚铁酸盐[28]。反应过程中,酚被氧化成苯氧自由基,根据反应条件的不同,可以进一步通过自由基耦合反应产生聚合、或重排产生苯醌、或导致烷基-芳香基断裂、Cα的氧化以及Cα−Cβ和芳香环的断裂[29]。Xu比较了若干漆酶对一系列苯酚类、苯胺类和苯硫酚类底物的催化反应,发现催化效率与底物结构以及Cu1和底物间单电子氧化还原电势差密切相关[30],而当底物在o-和p-位置同时存在基团时,T. trogii漆酶催化底物反应效率会明显增加。因此,底物的原子空间排布和氧化还原电势与漆酶催化反应效率密切相关[24]。
漆酶不能直接氧化具有高氧化还原电势的底物,但一些白腐真菌漆酶在木质素降解中发挥着重要作用。一些相应的化合物 (称为介体mediator) 作为漆酶的中介底物,使漆酶可以间接催化大分子底物和非芳香族化合物的氧化反应[31]。针对纸浆去木质素的漆酶/介体系统 (LMS) 使用的第一个介体是ABTS,之后大约有100多个化合物用作介体来测试氧化木质素的能力。一个有效的介体应该是一个合适的漆酶底物,它氧化后的自由基具有较长的半衰期,并带有较高的氧化电位。N-O·自由基在活性和稳定性方面比苯氧自由基显得更加平衡[32],因此木质素降解中最有效的介体都含有N−OH基团的氮杂环化合物,特别是1-羟基苯并三唑 (HBT)。
Jeon等比较了在含有不同介体组成的“鸡尾酒”配方下 G. lucidum漆酶氧化五氯苯酚 (PCP) 的能力,发现“鸡尾酒”混合物比单个介体更能提高漆酶氧化PCP的能力。相比较香草醛和乙酰香兰酮,G. lucidum漆酶更倾向于利用 ABTS作为介体,但ABTS自由基和香草醛 (或乙酰香兰酮) 的同时存在可以显著地加快PCP的氧化并伴随着电子从香草醛 (或乙酰香兰酮) 到 ABTS自由基的转移[33]。这些结果显示香草醛和乙酰香兰酮介导ABTS和PCP之间的反应是通过漆酶和这些介体之间连续的电子传递而实现的。
漆酶具有宽泛的底物氧化能力,并能够利用氧分子作为电子受体,因此作为生物催化剂被广泛地应用于多个领域。在纸浆去木质素、生物漂白、工业废水净化、纺织染色工业、饮料果汁加工以及生物传感器和生物电池的加工等领域都具有很大的潜在应用价值[34]。近年来的若干研究集中在如何利用漆酶对工业染料进行降解,大部分工作都利用了可用的商业化染料作为模型污染物[35],并取得了很好的效果,甚至对皮革工业中使用的偶氮染料,漆酶亦具有较好的脱色效果[36]。从P. ostreatus中制备的漆酶混合物甚至可以对RBBR蒽醌染料进行脱色反应并达到70%的效果,当该制备物被固定化后还可以重复使用[37]。然而,真正的工业废水常常包括多种染料,对于混合的染料降解取得的数据还比较有限。这也是今后漆酶在应用领域的一个研究热点。目前,利用漆酶对模拟的纺织工业废水染料混合物的降解作用已经在 SOPHIED EU计划 (NMP2-CT-2004-505899,6FP) 框架下进行了成功的测试,标志着这项技术在纺织染料废水处理中实际应用的可能性。
漆酶亦可应用于化学合成。例如漆酶催化反应可用来将儿茶酚单体聚合成聚邻苯二酚或产生惰性苯酚类聚合物 (如多聚萘酚)[38]。这些聚合物可以用于复合木材、纤维焊接、薄片材料、涂料和粘合剂的制备。而利用漆酶催化的交联反应原理设计的一个酶促聚合反应系统可以用来生产人造漆聚合膜(日本传统涂料)。漆酶也被用于染料合成和制药业,如制备抗癌剂actinocin或植物抗毒素resveratrol[39]。
此外,合适的LMS系统能够很有效地降解芳香族化合污染物、漂白纸浆、控制树脂障碍以及进行染料脱色[40]。但利用工业合成这些介体并不可取,因为成本较高并且可能会产生有毒物质。天然介体的使用更有利于LMS系统的应用。事实上,一些木质素衍生的苯酚类化合物已经被证实可以介导漆酶的染料脱色、纸浆木质素去除和PAH的氧化作用,比如天然对香豆酸 (对羟基苯基丙烯酸) 作为漆酶介体在降解 3,4-苯并芘时比合成的介体具有更高的效率[41]。
3 展望
对于漆酶的研究与应用已有一百多年的历史,特别是现代光谱学、晶体学和结构量子化学等方法的应用极大地推动了我们对漆酶的结构和催化机理的了解,进而对漆酶在各个领域的应用具有建设性的指导意义。然而,对漆酶的研究和了解远没有达到预期的程度,还存在着诸如糖基如何发挥作用、电子如何在残基之间传递等难题有待解决,同时如何通过蛋白质工程来改造漆酶,进一步提高其实际应用范围也是漆酶研究中的一个焦点。这些也为我们今后研究漆酶提供了丰富的素材,而未来更多更高分辨率漆酶结构的解析以及各种新技术新方法的引入则是解答这些难题的关键。
总之,漆酶是一个古老的酶蛋白,具有很广阔的应用前景。无论是作为一个结构与功能关系的研究模型,还是在不久的将来作为绿色环保工具(Green tool) 应用于生物技术工业,都具有非常巨大的实用性价值。
REFERENCES
[1] Solomon EI, Sundaram UM, Machonkin TE. Multicopper oxidases and oxygenases. Chem Rev, 1996, 96(7):2563−2606.
[2] Baldrian P. Fungal laccases-occurrence and properties.FEMS Microbiol Rev, 2006, 30(2): 215−242.
[3] Bao W, O'Malley D M, Whetten R, et al. A laccase associated with lignification in Loblolly Pine Xylem.Science, 1993: 260(5108): 672−674.
[4] Enguita FJ, Martins LO, Henriques AO, et al. Crystal structure of a bacterial endospore coat component − A laccase with enhanced thermostability properties. J Biol Chem, 2003, 278(21): 19416−19425.
[5] Shleev SV, Morozova OV, Nikitina OV, et al. Comparison of physico-chemical characteristics of four laccases from different basidiomycetes. Biochimie, 2004, 86(9/10):693−703.
[6] Palmer AE, Lee SK, Solomon EI. Decay of the peroxide intermediate in laccase: reductive cleavage of the O-O bond. J American Chemical Society, 2001, 123(27):6591−6599.
[7] Garavaglia S, Cambria MT, Miglio M, et al. The structure of Rigidoporus lignosus Laccase containing a full complement of copper ions reveals an asymmetrical arrangement for the T3 copper pair. J Mol Biol, 2004,342(5): 1519−1531.
[8] Kumar SV, Phale PS, Durani S, et al. Combined sequence and structure analysis of the fungal laccase family.Biotechnol Bioeng, 2003, 83(4): 386−394.
[9] Ge H, Gao Y, Hong Y, et al. Structure of native laccase B from Trametes sp. AH28-2. Acta Crystallogr Sect F Struct Biol Cryst Commun, 2010, 66(Pt 3): 254−258.
[10] Ducros V, Brzozowski AM, Wilson KS, et al. Crystal structure of the type-2 Cu depleted laccase from Coprinus cinereus at 2.2 A resolution. Nat Struct Biol, 1998, 5(4):310−316.
[11] Piontek K, Antorini M, Choinowski T. Crystal structure of a laccase from the fungus Trametes versicolor at 1.90-A reso-lution containing a full complement of coppers. J Biol Chem, 2002, 277(40): 37663−37669.
[12] Bertrand T, Jolivalt C, Briozzo P, et al. Crystal structure of a four-copper laccase complexed with an arylamine:insights into substrate recognition and correlation with kinetics. Biochemistry, 2002, 41(23): 7325−7333.
[13] Ferraroni M, Myasoedova NM, Schmatchenko V, et al.Crystal structure of a blue laccase from Lentinus tigrinus:evi-dences for intermediates in the molecular oxygen reductive splitting by multicopper oxidases. BMC Struct Biol, 2007, 7: 60.
[14] Matera I, Gullotto A, Tilli S, et al. Crystal structure of the blue multicopper oxidase from the white-rot fungus Trametes trogii complexed with p-toluate. Inorganica Chimica Acta, 2008, 361(14/15): 4129−4137.
[15] Polyakov KM, Fedorova TV, Stepanova EV, et al.Structure of native laccase from Trametes hirsuta at 1.8 A resolution. Acta Crystallogr D Biol Crystallogr, 2009,65(Pt 6): 611−617.
[16] Hakulinen N, Kiiskinen LL, Kruus K, et al. Crystal structure of a laccase from Melanocarpus albomyces with an intact trinuclear copper site. Nat Struct Biol, 2002,9(8): 601−605.
[17] Hakulinen N, Andberg M, Kallio J, et al. A near atomic resolution structure of a Melanocarpus albomyces laccase.J Struct Biol, 2008, 162(1): 29−39.
[18] DeLano WL. The PyMOL molecular graphics system.http://wwwpymolorg 2002.
[19] Lee SK, George SD, Antholine WE, et al. Nature of the intermediate formed in the reduction of O(2) to H(2)O at the trinuclear copper cluster active site in native laccase. J American Chemical Society, 2002, 124(21): 6180−6193.
[20] Solomon EI, Augustine AJ, Yoon J. O2reduction to H2O by the multicopper oxidases. Dalton Trans, 2008(30):3921−3932.
[21] Xu F, Shin W, Brown SH, et al. A study of a series of recombinant fungal laccases and bilirubin oxidase that exhibit sig-nificant differences in redox potential,substrate specificity, and stability. Biochim Biophys Acta,1996, 1292(2):303−311.
[22] Gray HB, Malmstrom BG, Williams RJ. Copper coordination in blue proteins. J Biol Inorg Chem, 2000,5(5): 551−559.
[23] Xu F. Effects of redox potential and hydroxide inhibition on the pH activity profile of fungal laccases. J Biol Chem,1997, 272(2): 924−928.
[24] Tadesse MA, D'Annibale A, Galli C, et al. An assessment of the relative contributions of redox and steric issues to lac-case specificity towards putative substrates. Org Biomol Chem, 2008, 6(5): 868−878.
[25] Torres J, Svistunenko D, Karlsson B, et al. Fast reduction of a copper center in laccase by nitric oxide and formation of a peroxide intermediate. J American Chemical Society,2002, 124(6): 963−967.
[26] Quintanar L, Yoon J, Aznar CP, et al. Spectroscopic and electronic structure studies of the trinuclear Cu cluster active site of the multicopper oxidase laccase: nature of itscoordination unsaturation. J American Chemical Society,2005, 127(40): 13832−13845.
[27] Yoon J, Liboiron BD, Sarangi R, et al. The two oxidized forms of the trinuclear Cu cluster in the multicopper oxidases and mechanism for the decay of the native intermediate. Proc Natl Acad Sci USA, 2007, 104(34):13609−13614.
[28] Giardina P, Palmieri G, Scaloni A, et al. Protein and gene structure of a blue laccase from Pleurotus ostreatus1. The Bio-chemical J, 1999, 341(Pt 3): 655−663.
[29] Kawai S, Umezawa T, Higuchi T. Degradation mechanisms of phenolic beta-1 lignin substructure model compounds by laccase of Coriolus versicolor. Archives Biochem Biophy, 1988, 262(1): 99−110.
[30] Xu F. Oxidation of phenols, anilines, and benzenethiols by fungal laccases: correlation between activity and redox po-tentials as well as halide inhibition. Biochemistry,1996, 35(23): 7608−7614.
[31] Bourbonnais R, Paice MG. Oxidation of non-phenolic substrates. An expanded role for laccase in lignin biodegradation. FEBS Lett, 1990, 267(1): 99−102.
[32] Xu F, Kulys JJ, Duke K, et al. Redox chemistry in laccase-catalyzed oxidation of N-hydroxy compounds.Appl Environ Microbiol, 2000, 66(5): 2052−2056.
[33] Jeon JR, Murugesan K, Kim YM, et al. Synergistic effect of laccase mediators on pentachlorophenol removal by Gano-derma lucidum laccase. Appl Microbiol Biotechnol,2008, 81(4): 783−790.
[34] Abadulla E, Tzanov T, Costa S, et al. Decolorization and detoxification of textile dyes with a laccase from Trametes hirsuta. Appl Environ Microbiol, 2000, 66(8): 3357−3362.
[35] Pereira L, Coelho AV, Viegas CA, et al. Enzymatic biotransformation of the azo dye Sudan Orange G with bacterial CotA-laccase. J Biotechnol, 2009, 139(1):68−77.
[36] Couto SR. Decolouration of industrial azo dyes by crude laccase from Trametes hirsuta. J Hazardous Materials,2007, 148(3): 768−770.
[37] Palmieri G, Giardina P, Sannia G. Laccase-mediated remazol Brilliant Blue R decolorization in a fixed-bed bioreactor. Biotechnol Prog, 2005, 21(5): 1436−1441.
[38] Ceylan H, Kubilay S, Aktas N, et al. An approach for prediction of optimum reaction conditions for laccase-catalyzed bio-transformation of 1-naphthol by response surface methodology (RSM). Biores Technol,2008, 99(6): 2025−2031.
[39] Ossiadacz J, Al-Adhami A, Bajraszewska D, et al. On the use of Trametes versicolor laccase for the conversion of 4-methyl-3-hydroxyanthranilic acid to actinocin chromophore. J Biotechnol, 1999, 72: 141−149.
[40] Gutierrez A, Del Rio JC, Ibarra D, et al. Enzymatic removal of free and conjugated sterols forming pitch deposits in environmentally sound bleaching of eucalypt paper pulp. Environ Sci & Technol, 2006, 40(10): 3416−3422.
[41] Canas A, Alcalde M, Plou FJ, et al. Transformation of polycyclic aromatic hydrocarbons by laccase is strongly enhanced by phenolic compounds present in soil. Environ Sci & Technol, 2007, 41: 2964−2971.
Technol, 2005, 16(8): 344−350.
Structure, catalytic mechanism and applications of laccases: a review
Honghua Ge, Yun Wu, and Yazhong Xiao
Modern Experiment Technology Center & School of Life Sciences, Anhui University, Hefei 230039, China
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
天津城建大学学报(2018年6期)2019-01-15 05:51:40
安徽农学通报(2017年8期)2017-05-12 10:45:07
农业与技术(2015年3期)2016-01-18 03:18:24
池州学院学报(2015年3期)2016-01-05 01:13:04
合成生物学(2015年5期)2015-12-19 07:10:22
电源技术(2015年5期)2015-08-22 11:18:08
石油化工(2015年11期)2015-08-15 00:43:05
天津科技大学学报(2015年2期)2015-08-09 01:40:42
