
利用分子标记早期筛选光皮桦核心种质
2011-07-30 10:58李海英梁志伟黄华宏童再康卢泳全
浙江林业科技 2011年3期
李海英,梁志伟,陈 冲,黄华宏,童再康,卢泳全
(浙江农林大学 亚热带森林培育国家重点实验室培育基地,浙江 临安 311300)
植物遗传资源是物种多样性保护及品种选育的重要物质基础。理论上保存的遗传资源越多越好,但种质资源保存过多会耗费巨大的人力物力,也难以对所保存的材料进行合理评价,制约了种质的利用。因此,Frankel于1984年提出了核心种质(core collection)的概念[1],即以最少数量的种质材料代表一个物种及其近缘野生种最大限度的遗传多样性。核心库中入选的遗传材料都要有代表性,并包括尽可能多的遗传多样性。
构建核心种质库主要涉及两方面的内容,一是构建核心种质所采用的抽样方法;二是核心种质遗传多样性的评价指标。在传统的方法中,一般是以形态学及农艺性状为评价指标[2~4]。但是,由于木本植物形态学性状观察周期长,与经济价值相关的材性性状往往需要几十年的时间才能准确观测,因而制约了多年生木本植物核心种质库的构建。目前,只在白桦[5]、腊梅[6]和枣[7]等几个物种中有相关报道。
近年来,随着生物技术的迅猛发展,分子标记越来越多地应用到种质资源研究中。由于分子标记是以个体间遗传物质—核苷酸序列变异为基础的遗传标记,是DNA水平遗传变异的直接反映。因此,在生物发育的不同阶段,不同组织的DNA都可用于遗传资源的标记分析[8]。由于分子标记所揭示的多态性不受外界环境和内在发育阶段的影响,所以非常适于多年生木本植物种质资源的早期/幼苗期鉴定。刘勇等[9]利用分子标记技术确立了柚的核心种质资源。研究结果表明,核心种质能代表初始种质群。
光皮桦(Betula luminifera)是我国南方山地常见的优良速生用材阔叶树种,耐贫瘠,生长快,材质好,是一种极具推广价值的造林树种。本研究于2005年和2006年分别从浙江、广西、福建、贵州四省的9个光皮桦天然种源采集种子,温室内种植。实生苗生长1 a后,采优株穗条嫁接于浙江农林大学光皮桦种质园。目前,该种子园共有材料519份。本研究旨在利用分子标记技术,从这些资源中筛选具有代表性的核心资源,为光皮桦种质资源的合理保存及早期利用奠定理论基础。
1 材料与方法
1.1 初始种质的选取
本实验采用分组取样法,根据种源差异将整个群体分为9个组(见表1)。然后组内采用随机取样,结合组内个体的表现型差异大小确定各组的取样比例。最终,从519份共9组光皮桦样本中抽取62份作为初始种质。于2009年5月,取正常生长的嫩叶,采用CTAB+硅珠吸附法[10]提取叶片总DNA。
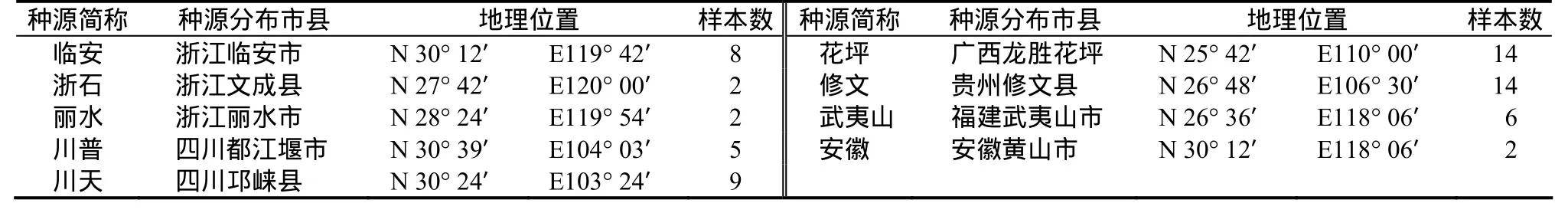
表1 光皮桦种源地理位置Table 1 Geographic location of samples
1.2 分子标记方法
采用由本实验室开发的扩增共有序列遗传标记(Amplified Consensus Genetic Markers, ACGM)分子标记[11]对光皮桦进行遗传多样性分析,共得到137个多态性位点。以1和0记录等位基因的有和无,获得矩阵。利用系统分析软件POPGENE 32计算居群的多态位点数、多态位点百分率(percentage of polymorphic belt, PPB%)、观测等位基因数(Na)、有效等位基因数(Ne)、Nei基因多样性指数和 Shannon信息指数[9],按类平均数聚类方法(UPGMA)进行聚类,得到UPGMA系统树。
1.3 柚类核心种质构建方法
利用ACGM所得到的谱带进行聚类分析,按照聚类分析结果并结合形态学性状,将遗传相似系数最大的2个种质删除1份,剩余的种质再聚类;再从遗传相似系数最大的成对种质中删除一份。以此类推,直到代表性或核心种质量达到要求。利用上述方法分别抽取41、36、27、18和9个样品组成核心样本群进行分析比较,各样品群代号分别为I、II、III、IV、V。
样品群 I(41 个样本)代号为:2,3,4,5,6,7,9,10,11,12,13,14,15,16,17,18,20,21,22,26,27,28,30,31,37,38,41,43,44,46,47,48,50,57,58,59,60,61,62,64,65。
样品群 II(36 个样本)代号为:3,4,5,6,7,9,10,11,12,13,14,15,16,17,18,20,21,26,27,30,31,37,38,41,43,44,46,47,48,58,59,60,61,62,64,65。
样品群 III(27 个样本)代号为:4,5,6,9,10,11,14,15,16,17,18,21,27,30,31,41,43,44,48,58,59,62,64,65。
样品群IV(18个样本)代号为:4,5,9,10,11,14,15,17,18,27,30,31,43,44,58,59,64,65。
样品群V(9个样本)代号为:5,10,14,17,27,30,44,58,64。
1.4 核心种质的评价
构建好核心种质后,用多态位点数、多态位点百分率、观测等位基因数、有效等位基因数、Nei基因多样性指数和Shannon信息指数评价指标来评价核心种质。利用Microsoft Excel中的分析数据库,对初始种质和核心种质的观测等位基因数、有效等位基因数、Nei基因多样性指数和Shannon信息指数进行t检验[9],根据检验结果来评价核心种质。
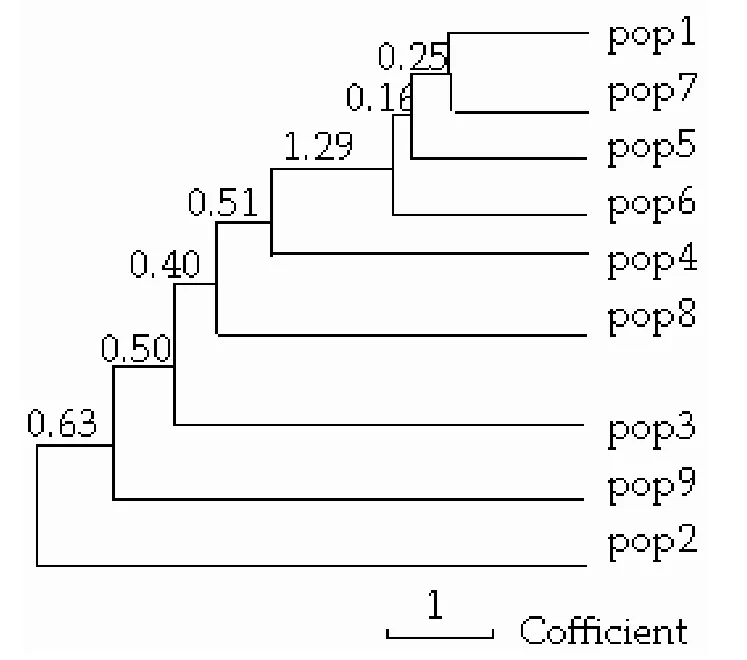
图1 9个居群UPGMA聚类Figure 1 UPGMA dendrogram among 9 populations
2 结果与分析
2.1 初级种质的遗传多样性分析
通过PopGen32[9]软件分析得到居群聚类结果(图1),临安(pop1)和修文(pop7)最先聚在一起,然后川天(pop5)、花坪(pop6)、川普(pop4)、武夷山(pop8)、丽水(pop3)、安徽(pop9)和浙石(pop2)依次相聚。从光皮桦种源的地理分布上看,修文(pop7)、川天(pop5)、花坪(pop6)和川普(pop4)位于我国西南地区的贵州、四川和广西三省,而武夷山(pop8)、丽水(pop3)、安徽(pop9)和浙石(pop2)来源于我国东南地区的福建、浙江和安徽三省。由此可见,本研究的聚类结果与光皮桦各种源的地理分布是相符的。
2.2 各样本群的遗传多样性比较
利用 PopGen32软件分析各样本群的多态性位点数、多态性位点百分率、观测等位基因数、有效等位基因数、Nei遗传多样性指数和Shannon信息指数(结果见表2)。比较不同取样数目所获得的各种遗传参数,结果表明,样品群II的有效等位基因数是最高的,多态性位点数、多态性位点百分率、观测等位基因数、Nei遗传多样性指数和Shannon信息指数等仅比样品群I的稍低,考虑到取样数的问题,我们把样本群II列为核心种质。最后获得的核心种质的编号为:3,4,5,6,7,9,10,11,12,13,14,15,16,17,18,20,21,26,27,30,31,37,38,41,43,44,46,47,48,58,59,60,61,62,64,65。

表2 各个样品群的遗传多样性分析Table 2 Analysis of genetic diversities among different sampling groups
2.3 核心种质的评价
用 PopGen32软件比较初始种质与核心种质的观测等位基因数、有效等位基因数、Nei遗传多样性指数、Shannon 信息指数和多态性位点百分率(结果见表3),并对两个群体的各参数进行t测验(结果见表4)。从表3可看出核心种质保留了初始种质58.06%的样品,多态性位点保留率达到了96%,观测等位基因数、有效等位基因数、Nei遗传多样性指数和Shannon信息指数的保留率分别为 98.58%、99.88%、98.74%、98.30%。由 t测验的结果可表明,核心种质的观测等位基因数、有效等位基因数、Nei遗传多样性指数和Shannon信息指数在概率0.01水平上与初始种质差异不显著,核心种质能很好的代替初始种质。

表3 初始种质与核心种质遗传多样性对比Table 3 Comparision of the genetic diversities between primary sample and core collection

表4 初始种质与核心种质t测验结果Table 4 t-test result between initial collection and core collection
3 讨论
在构建核心种质时,抽样比例很重要。目前,多数植物核心种质的抽样比例为全部收集种质的5% ~ 30%,一般为10%左右[12]。但是因为生物进化及人工选择对作物干预的存在,产生了各个物种的特性,所以对整体的取样比例不能简单格式化,应视研究作物的遗传结构及遗传多样性而定。在本研究中,构建初始种质时,抽样比例为12%,与多数植物核心种质的抽样比例适当。但是在构建核心种质时,因为光皮桦初始种质的样本数量较少,只有62份,所以得到的核心种质的比例为初始种质的58.06%。
抽样技术也很重要,直接关系到核心库的好坏。常用的组内取样法有按分层抽样[13]、遗传差异取样[14]、组内随机取样[15]、多次聚类抽样[16]等方法。本实验中采用组内随机取样,先按聚类分析分组,再从各组随机抽取样品得到光皮桦核心种质,最后通过 t检验,证明本研究得到的核心种质能很好的代表原光皮桦资源群体的遗传变异和遗传结构。
[1]Frankel O H. Genetic Manipulation : Impact on Man And Society[M]. Cambridge Uni Press,1984. 161-170.
[2]Brown A H D. The use of plant genetic resources [A]. Brown A H D,Frandel O H,Marshall R D,et al. [C]. England: Cambridge Univ Press,1989.
[3]Corley Holbrook C, William F Anderson. Evaluation of a core collection to identify resistance to late leafspot in peanut[J]. Core Science,1995(35):1700-1702.
[4]沈金雄,郭庆元,张秀荣,等. 中国芝麻种质资源的聚类分析[J]. 华中农业大学学报,1995,14(6):532-536.
[5]魏志刚,高玉池,刘桂丰,等. 白桦核心种质的初步构建[J]. 林业科学,2009,45(10):74-78.
[6]赵冰. 腊梅种质资源遗传多样性与核心种质构建的研究[D]. 北京:北京林业大学,2008.
[7]董玉慧. 枣树农艺性状遗传多样性评价与核心种质构建[D]. 石家庄:河北农业大学,2008.
[8]李丽,何伟明,马连平,等. 用EST-SSR分子标记构建大白菜核心种质及其指纹图谱[J]. 基因组学与应用生物学,2009,28(1):76-88.
[9]刘勇,孙中海,刘德春,等. 利用分子标记技术选择柚类核心种质资源[J]. 果树学报,2006,23(3):339-345.
[10]张博,张露,诸葛强,等. 一种高效的树木总DNA的提取方法[J]. 南京林业大学学报(自然科学版),2004,28(1):13-15.
[11]Yongquan Lu,Haiying Li,Xi Chen,et al. Development amplication concensus genome markers in Betula luminifera based on Birch EST database[J]. Wood Res, 2011, 56(2):169-178.
[12]李自超,张洪亮,曹永生,等. 中国地方稻种资源初级核心种质取样策略研究[J]. 作物学报,2003,29(1):20-24.
[13]刘三才,郑殿升,曹永生,等. 普通小麦核心种质抽样方法的比较[J]. 麦类作物学报,2001,21(2):42-45.
[14]徐海明,胡晋,朱军. 构建作物种质资源核心库的一种有效抽样方法[J]. 作物学报,2000,26(2):157-162.
[15]魏兴华,颜启传,应存山,等. 建立浙江地方籼稻种资源的核心样品研究[J]. 中国水稻学,1999,13(2):81-85.
[16]HU J,Zhu J,Xu H M. Methods of constructing core coolletions by stepwise clustering with three sampling strategies based on the genotypic values of crops[J]. Theor Appl Genet,2000(101):264-268.
猜你喜欢
今日农业(2022年14期)2022-09-15
今日农业(2022年13期)2022-09-15
世界科学技术-中医药现代化(2022年3期)2022-08-22
川北医学院学报(2022年6期)2022-06-24
智慧健康(2021年17期)2021-07-30
昆明医科大学学报(2021年3期)2021-07-22
中国产前诊断杂志(电子版)(2020年1期)2020-05-21
四川蚕业(2020年4期)2020-02-10
遵义医科大学学报(2020年6期)2020-02-05
中国麻业科学(2018年6期)2018-04-09
