
高效液相色谱法测定复方萘甲唑啉鼻喷雾剂含量
2011-05-17 09:56周建林杜长生
药学与临床研究 2011年4期
周建林,杜长生
南京星银药业有限公司,南京 211200
复方萘甲唑啉鼻喷雾剂主要含有萘甲唑啉和马来酸氯苯那敏,两者分别为抗组胺和血管收缩药,对鼻黏膜充血、肿胀有较好的疗效。适用于急性鼻炎、鼻窦炎以及变态反应性鼻炎的发作期。目前含盐酸萘甲唑啉和马来酸氯苯那敏的复方制剂,有滴鼻液、滴眼液等,对两药的含量测定,部颁标准[1]采用分光光度法,本文则综合考察文献方法[2-5]建立高效液相色谱法,测定该剂型两成分含量。
1 仪器和试药
LC-10ATVP 高效液相色谱仪 (日本岛津),SPD-10AVP紫外检测器,CTO-10AVP柱温箱,浙大N-2000色谱工作站。
复方萘甲唑啉鼻喷雾剂样品 (规格:10 mL,盐酸萘甲唑啉5 mg,马来酸氯苯那敏10 mg)批号为090601、090701、090702、090703、090704,由南京星银药业有限公司生产;盐酸萘甲唑啉对照品、马来酸氯苯那敏对照品均由中国药品生物制品检定所提供;甲醇为色谱纯;三乙胺、磷酸二氢钾、磷酸均为分析纯;水为重蒸馏水。
2 方法和结果
2.1 色谱条件与系统适用性试验
色谱柱为汉邦 C18柱(4.6 mm×250 mm,5 μm);流动相:甲醇-水-三乙胺-磷酸二氢钾-磷酸(540︰460︰2︰3.4︰1);流速 1 mL·min-1;柱温 30℃;紫外检测波长264 nm;进样量20 μL。理论塔板数按萘甲唑啉峰计算不低于3000;按氯苯那敏峰计算不低于3000。萘甲唑啉和氯苯那敏的分离度大于1.5。主药、杂质呈基线分离。液相色谱图见图1。

图1 盐酸萘甲唑啉、马来酸氯苯那敏的液相色谱图
2.2 溶液制备
2.2.1 对照品溶液精密称取经105℃干燥至恒重的盐酸萘甲唑啉对照品25 mg,马来酸氯苯那敏对照品50 mg,置50 mL量瓶中,加水溶解并稀释至刻度,摇匀;再精密量取5 mL,置100 mL量瓶中,加水稀释至刻度,摇匀,作为对照品溶液。
2.2.2 供试品溶液精密量取供试品5mL置100mL量瓶中,加水稀释至刻度,摇匀,作为供试品溶液。
2.3 线性关系考察
精密称取马来酸氯苯那敏对照品20 mg和盐酸萘甲唑啉对照品10 mg,置于50 mL量瓶中,加水适量使溶解,后加水稀释至刻度,摇匀,制成混合对照溶液。分别精密吸取上述混合对照溶液1、2、3、5、10 mL分别置50 mL量瓶中,用流动相稀释至刻度,摇匀,分别精密吸取上述5种浓度对照液20 μL注入液相色谱仪,测定峰面积,以进样浓度X(μg·mL-1)为横坐标,以峰面积Y为纵坐标,绘制标准曲线,线性回归方程分别为:马来酸氯苯那敏:Y=1.84×104X+3.45×103,r=1.0000; 盐酸萘甲唑啉:Y=2.75×104X-1.38×104,r=0.9976。 结果表明,马来酸氯苯那敏浓度在 8.03~80.30 μg·mL-1和盐酸萘甲唑啉浓度在4.04~40.40 μg·mL-1线性关系良好。
2.4 精密度试验
分别精密吸取复方萘甲唑啉鼻喷雾剂供试品溶液,进样20 μL,按上述色谱条件连续重复进样5次,测定峰面积,RSD为0.87%。
2.5 重复性试验
取复方萘甲唑啉鼻喷雾剂供试品按上述供试品溶液的制备方法,平行制备5份供试品溶液,进样20 μL,计算盐酸萘甲唑啉的含量为98.9%,RSD=0.9%,马来酸氯苯那敏的含量为97.8%,RSD=1.2%。
2.6 回收率试验
取复方萘甲唑啉鼻喷雾剂供试品5份,分别精密吸取5mL,置于100mL量瓶中,精密加入盐酸萘甲唑啉对照品溶液和马来酸氯苯那敏对照品溶液各5mL,用水溶解稀释至刻度,进样20 μL,测得回收率为98.22%~100.13%,n=5,RSD<0.85%。分别称取盐酸萘甲唑啉和马来酸氯苯那敏的对照品,配制成浓度为0.6 mg·mL-1和 1.2mg·mL-1、0.5mg·mL-1和 1.0mg·mL-1、0.4 mg·mL-1和 0.8mg·mL-1,考察了高、中、低 3 个剂量的回收率,分别为98.58%、99.14%、100.43%。
2.7 稳定性考察
取供试品溶液在 0、2、4、6、8、10、12 h 测定峰面积,计算RSD=1.7%。表明供试品在12 h内稳定。
2.8 最小检测限
取马来酸氯苯那敏对照品溶液 (浓度0.04 mg·mL-1),逐级稀释,直至信噪比(S/N)=3,最小检测浓度为 2 ng·mL-1,检测限度为 0.04 ng(2 ng·mL-1×20 μL)。
取盐酸萘甲唑啉对照品溶液 (浓度0.05mg·mL-1),逐级稀释,直至信噪比(S/N)=3,最小检测浓度为12.5 ng·mL-1,检测限度为 0.25ng(12.5ng·mL-1×20μL)。
2.9 含量测定
5个不同批号的供试品分别用高效液相色谱法与分光光度法测定标示含量,结果见表1。
3 讨 论
本实验考察了流动相中不同比例甲醇-水、甲醇-磷酸二氢钾溶液体系对测定的影响,在甲醇-水为流动相时,马来酸氯苯那敏出现分裂峰,说明流动相比例和流动相的pH值不适宜,造成马来酸氯苯那敏的分裂峰,出现了马来酸和氯苯那敏两个峰;当改变流动相条件,加入磷酸二氢钾溶液和三乙胺,能改善马来酸氯苯那敏的峰形和解决盐酸萘甲唑啉、马来酸氯苯那敏峰形拖尾现象。合适的磷酸二氢钾溶液浓度能避免色谱柱的堵柱现象。本法在进行辅料干扰试验时,未发现辅料对测定结果的干扰。本法能够有效地控制产品的质量。
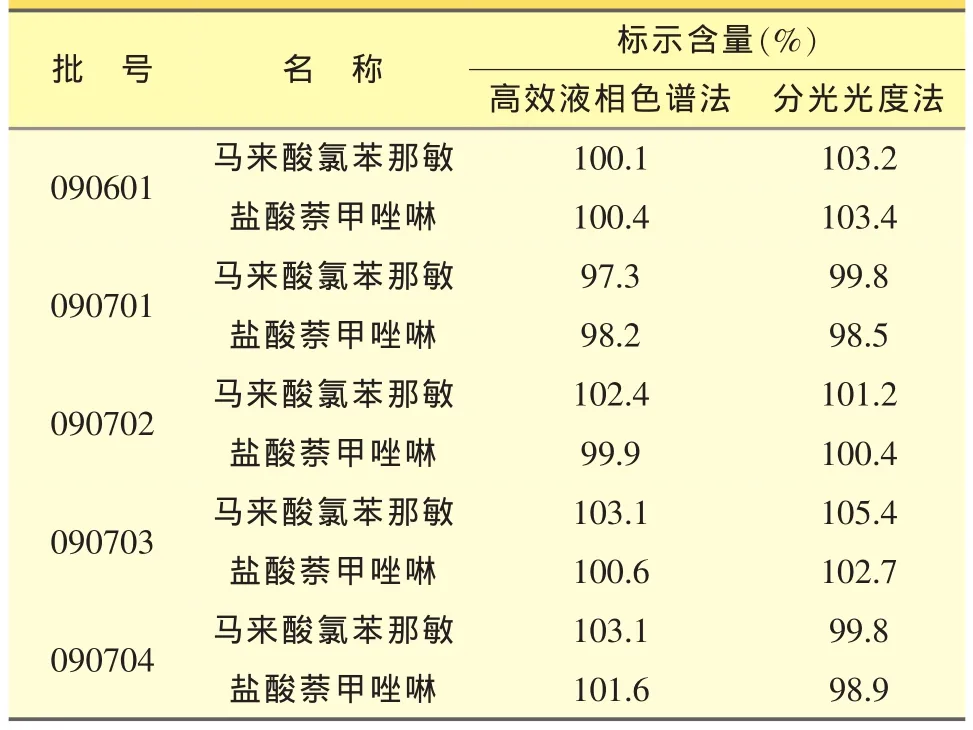
表1 高效液相色谱法与分光光度法测定结果
[1] 国家药典委员会.卫生部药品标准:新药转正标准第十四册[S].北京:人民卫生出版社,1998:12-4.
[2] 袁宇琳,冉 兰,刘梅妍,等.HPLC测定复方萘甲唑啉鼻喷剂中的有关物质 [J].华西药学杂志,2009,24(1):88-9.
[3] 白若琬,潘继飞,朱建东,等.HPLC测定萘敏己锌滴眼液中盐酸萘甲唑啉和马来酸氯苯那敏含量[J].食品与药品,2010,12(7):238-41.
[4] 李国成,刘春霞,余晓霞,等.HPLC测定鼻炎灵滴鼻液中盐酸萘甲唑啉和马来酸氯苯那敏含量的方法研究[J].国际医药卫生导报,2010,14(2):192-6.
[5] 国家药典委员会.中华人民共和国药典:二部 [M].北京:中国医药科技出版社,2010:附录29-31.
猜你喜欢
化工进展(2022年9期)2022-10-13
中国美容医学(2022年3期)2022-04-27
四川大学学报(自然科学版)(2022年1期)2022-02-10
医学概论(2021年18期)2021-01-21
作文中学版(2019年11期)2019-11-27
家庭百事通·健康一点通(2019年6期)2019-07-08
作文中学版(2018年12期)2018-11-28
健康大视野(2018年10期)2018-10-29
上海预防医学(2017年1期)2017-03-09
科技与创新(2015年18期)2015-09-11
