
除菌过滤器完整性检测及风险评估
2011-02-27 02:27郑金旺
化工与医药工程 2011年4期
郑 军 郑金旺
(上海东富龙科技股份有限公司,上海 201108)
1 引言
除菌过滤器的完整性检测通常有以下四种方案:离线手动检测、离线自动检测、在线手动检测与在线自动检测。检测方案的每一步发展对药品生产可能产生的污染及交叉污染都在减少。但是,从检测方案的风险评估表中可以看出还是没有达到最大限度的降低风险。本文中提出的除菌过滤器在线完整性检测方案是在在线自动检测的基础上从软件与硬件两方面改进而来的。它能最大限度的降低整个检测过程对药品生产带来的风险。
2 过滤器完整性检测相关的规定
2.1 我国新版GMP
附录1《无菌药品》第四十二条 进入无菌生产区的生产用气体(如:压缩空气、氮气,但不包括可燃性气体)均应经过除菌过滤,应当定期检查除菌过滤器和呼吸过滤器的完整性。
附录1《无菌药品》第七十五条 非最终灭菌产品的过滤除菌应当符合以下要求,第三点 除菌过滤器使用后,必须采用适当的方法立即对其完整性进行检查并记录。常用的方法有起泡点试验、扩散流试验或压力保持试验。
2.2 欧盟GMP
应采用适当的方法对除菌过滤器在使用前的完好性进行检查,并在使用后立即确认其完好性。常用的方法有:起泡点、扩散流试验或压力保持试验。
2.3 PDA 技术报告NO.26 2008:第7节 完整性检测
建议使用前与过滤后(使用后)完整性检测。
2.4 FDA
正常情况下,过滤器应在过滤器前,及组装好后,且灭菌后执行完整性检测。
在过滤器后对过滤器进行完整性检测,从而查看过滤器是否泄露和膜已破损。
3 除菌过滤器的定义
FDA关于除菌级过滤器的定义:“一个除菌级过滤器必须是当以>107cfu/cm2假单胞菌(Brevundimonas diminuta) 进行挑战时下游滤出液无菌的过滤器。”而PDA的细菌截留试验使用每平方厘米5 ×108cfu B. diminuta 挑战0.2 µm 级的膜。图1所示为细菌截留试验。

图1 过滤器细菌截留试验
不管是哪种标准,只要过滤器能够在如此极端恶劣情况下滤出的无菌液体,就可以把这个过滤器称之为除菌过滤器,而不考虑它的孔径大小。
这种试验对于过滤器来说是破坏性试验,由过滤器供应商完成。在线使用的除菌过滤器不能使用这种方案检测其完整性。完整性检测只能是非破坏性的物理性检测。非破坏性的物理性检测数据必须能够与破坏性的细菌截留试验相关联。通常有前进流检测,起泡点检测,压力衰减,也可以进行前进流与起泡点的联合检测。
完整性检测可以人工实施,或使用完整性检测自动工具进行。根据是否使用完整性检测自动工具与是否在线,完整性检测有以下几种方案,一是离线手动检测,二是离线自动检测,三是在线手动检测,四是在线自动检测。下面四章将分别介绍这四种检测方案。
4 离线手动检测
图2所示的是离线手动前进流检测方案。前进流检测需要三个参数,检测压力,检测时间,最大流量。三个参数由过滤器供应商提供。检测前过滤器需要充分的湿润。上游关闭,在过滤器中通入可调的稳定气源。在一定的时间内,通过过滤器芯的测量扩散流气体体积与标准的数据比较,可以检测过滤器芯的完整性。
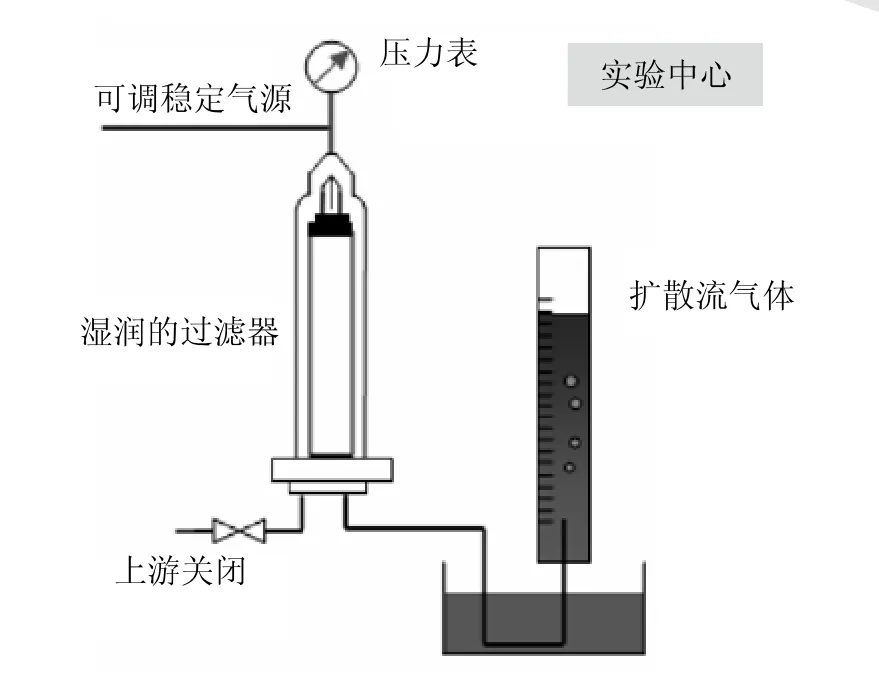
图2 离线手动前进流检测
相对完整性检测仪自动检测手动检测有以下缺点:敏感度低,结果的重复性低,下游的污染风险大。
5 离线自动检测
图3所示的是离线自动前进流检测方案。过滤器上游关闭,下游开口,过滤器芯充分的湿润。完整性检测进口接入可调气源,压力在仪器可承受范围内,在仪器面板输入检测参数,启动前进流检测即可。完整性检测仪不仅提供通入过滤器内的稳压的气源,也负责测量通过过滤器芯的扩散流气体体积。检测结束后,完整性检测仪根据设定的参数自动判断检测结果。
完整性检测仪有检测结果可打印存档,软件安全性好,操作简单,且有电子签名等优点。

图3 离线自动前进流检测
6 在线手动检测
图4所示的是在线手动前进流检测方案。
在线手动检测与离线自动检测的区别只有前者是在线检测,后者是下线检测。这里的手动指的是湿润过程与启动完整性检测手动。
在线检测与离线检测最大的区别:在线检测不仅能够检测过滤器的完整性也能够检测过滤器安装的完整性;在线检测降低了过滤器转移过程中的污染风险与损坏风险。
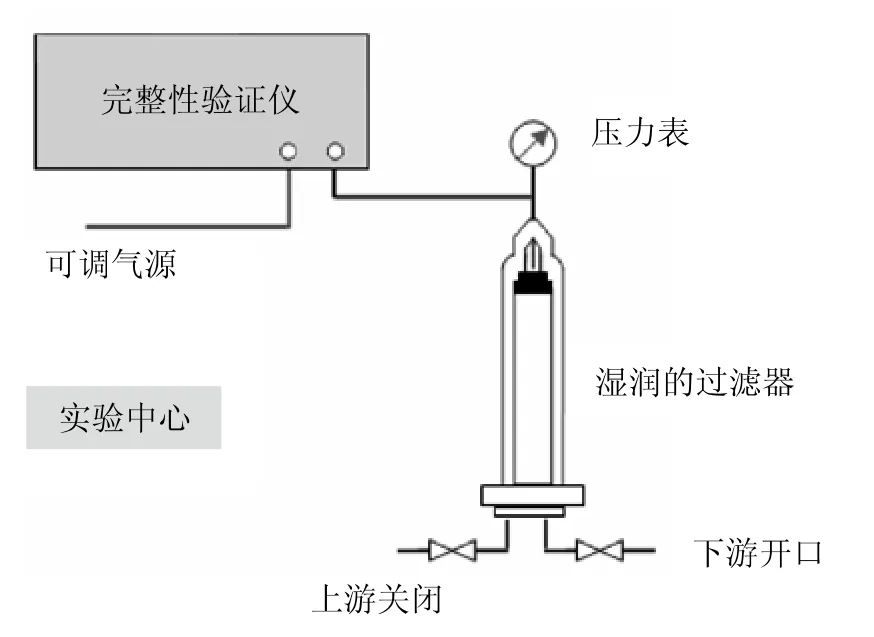
图4 在线手动前进流检测
7 在线自动检测
图5所示的是在线自动前进流检测方案。这个方案中,过滤器的湿润、参数的设定、检测的启动都是自动进行的,一键操作即可完成整个操作过程。
SCADA在系统中的作用是,参数的设定,历史数据的记录,检测的启动,过程的监控检测,结果的显示与记录。PLC的作用是,与完整性检测仪的通讯,检测流程的执行。

图5 在线自动前进流检测
湿润的过程是,过滤器湿润流程开始,打开过滤器湿润阀,打开溢流阀,湿润溶液在压缩空气的压力下进入过滤器。湿润用溶液可以通过液位计LS调整,浸泡时间也可以自由控制。浸泡完成后,湿润溶液通过排出阀排掉,即可进入检测阶段。
全自动控制的完整性检测较手动控制,有以下优点:操作更加简单;数据记录转入SCADA,保存时间更久,可记入批记录;湿润的过程减少人的操作,降低污染风险。
8 四种检测方案的风险评估
适用于生产用过滤器检测的以上四种方案,离线自动检测与在线手动检测应用最普遍。离线手动检测不仅有诸多弊端,而且不能进行电子签名与电子记录,基本上淘汰。而在线自动检测因为硬件的投入比较大,主要应用在高端冻干机上。
以上四种方案从无菌生产的角度考虑都有各自的缺点,是存在风险的检测方案,下面是四种检测方案的风险评估。

表1 过滤器完整性检测四种方案风险评估
为了降低过滤器的风险,我国新版GMP规定宜安装第二支过滤器。第一支过滤器是主过滤器,如果第一支过滤器检测合格,第二支过滤器就不需要检测;第一支过滤器检测不合格,第二支过滤器如果检测合格,还是承认过滤器的过滤效果,从而对本批药放行。两支过滤器在检测过程中对无菌性的要求不同,主要表现在过滤器上游的无菌性。第一支检测时对无菌性的要求比较低,第二支检测时对无菌性的要求比较高,因为第二支的检测会影响到第一支下游的无菌性。
所以,表1所述的方案的风险评估,尤其是在线检测的风险评估,针对第一支过滤器上游的所有风险项风险降低,而第二支过滤器上游的风险项风险则升高。
9 完善后的除菌过滤器在线完整性检测
通过表1可以看出在线自动检测的各项风险最低,即使如此还是有很多的风险项,为储溶液罐的消毒,湿润用溶液的无菌,进溶液管道的消毒,注入溶液到储罐的无菌操作,气体的无菌,进气管道,检测仪,下游开口。其中风险比较大的是下游开口,储罐的消毒,检测用介质的无菌。
我国新版GMP第一章总则第三条:本规范作为质量管理体系的一部分,是药品生产管理和质量控制的基本要求,旨在最大限度地降低药品生产过程中污染、交叉污染以及混淆、差错等风险,确保持续稳定地生产出符合预定用途和注册要求的药品。
基于以上原则,有必要通过改进系统降低除菌过滤器完整性检测中的风险。完善后的除菌过滤器在线完整性检测是在在线自动检测系统的基础上做些改进,包括硬件上与软件上的。
10 需要做的改进说明
10.1 针对每个风险项硬件上做以下修改,如表2。
10.2 软件上的修改
软件上主要是检测的流程及报警保护上需要做一些改进。比如首次检测失败,第二次检测时采用更大的压力,更长的时间湿润以排除湿润不当造成的检测失败。更多的报警与连锁,防止流程进入死循环,以确保检测能顺利的进行。

表2 硬件改进措施
11 结论
本文回顾了除菌过滤器完整性检测的四种方案,并且进行了风险评估,不管是哪种方案对无菌生产都有一定的风险,根据我国新版GMP的指导原则,有必要做些改进以确保最大限度的降低药品生产中的污染及交叉污染。
在第十章中,针对风险给出了一些建议。当然有些风险只能降低不能消除,比如检测仪带来的风险,因为检测仪本身的气体管道不能够消毒灭菌。如果在检测仪与过滤器之间增加设备截留细菌,首先要从理论上解决这种检测方案与细菌截留试验的数据关联。
即使有些风险不能消除,我们还是要努力采取措施,最大限度的降低风险,使除菌过滤器在线完整性检测对系统产生污染、交叉污染最大降低。
[1] 国家食品药品监督管理局 药品认证管理中心. 欧盟药品GMP指南[M]. 北京:中国医药科技出版社,2008.
[2] 国家食品药品监督管理局. 药品生产质量管理规范(2010年修订).
[3] 过滤器制药协会. 技术报告No. 26 (2008 修订版).
[4] 美国FDA. 无菌制剂生产质量管理规范(2004).
猜你喜欢
防爆电机(2021年4期)2021-07-28
中国特种设备安全(2021年11期)2021-05-05
哈尔滨轴承(2020年2期)2020-11-06
铁道通信信号(2020年6期)2020-09-21
电子制作(2019年7期)2019-04-25
电子制作(2018年19期)2018-11-14
电子制作(2018年9期)2018-08-04
中成药(2018年2期)2018-05-09
电子制作(2017年20期)2017-04-26
光学精密工程(2016年1期)2016-11-07
