
表达淀粉酶、糖化酶葡糖酸醋杆菌工程菌的构建
2011-01-11 12:36潘凌鸿李欣颜彩玲黄建忠
微生物学杂志 2011年6期
潘凌鸿,李欣,颜彩玲,黄建忠
(福建师范大学工业微生物教育部工程研究中心生命科学学院福建省现代发酵技术工程研究中心,福建福州350108)
细菌纤维素(Bacterial cellulose,简称BC),又称为β-1,4-葡聚糖,是某些细菌产生的一种细胞外多糖,它是由β-D-葡萄糖通过β-1,4-糖苷键连接而成的直链,直链之间无分支结构。与植物纤维素相比,细菌纤维素具有独特的优点,如高纯度(不掺杂半纤维素、木质素等)、良好的械性能、高持水性、高聚合度、高结晶度以及良好的生物亲和性和可降解性,不污染环境[1]。细菌纤维素已经应用于采油、纺织、生物医学、化工等行业,利用细菌纤维素制备创伤敷料、人造皮肤、软骨组织工程支架、哺乳动物细胞培养基质等是当前研究的热点[2]。在已经发现能产生纤维素的几个菌属中,以葡糖酸醋杆菌属(Gluconacetobacter),尤其是木葡糖酸醋杆菌(Gluconacetobacter xylinus)或汉氏葡萄酸醋杆菌(Gluconacetobacter hansenii)合成纤维素能力最强,它已经成为纤维素合成生化和遗传研究的模式生物[3]。2010年,G.hansenii ATCC23769全基因组测序完成,为研究该细菌提供了极大的便利[4]。葡糖酸醋杆菌以葡萄糖、D-果糖、D-甘露醇为碳源时纤维素产量较高[5]。但是由于这些单糖的成本过高,已成为细菌纤维素工业化生产和推广应用的瓶颈之一。在众多碳源中,淀粉来源广,易于获得,价格低廉,并且淀粉的水解产物葡萄糖分子可以直接作为合成纤维素的前体。但是由于葡糖酸醋杆菌本身不表达水解淀粉的酶类,在淀粉为碳源的培养基中不能产生细菌纤维素。本实验利用所构建内源表达载体,向G.hansenii ATCC23769体内导入枯草芽胞杆菌Bacillus subtilis WB600的淀粉酶基因、黑曲霉Aspergillus niger的糖化酶基因,以期使木醋杆菌在生长的同时,能够水解淀粉产生葡萄糖作为碳源,为降低细菌纤维素的生产成本创造可能的条件。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒汉氏葡糖酸醋杆菌Gluconacetobacter hanseniiATCC23769购于美国ATCC(American Type Culture Collection),枯草芽胞杆菌Bacillus subtilis WB600,大肠埃希菌E.coli DH5α由本实验室保存。能在E.coli和G.hansenii ATCC23769中自主复制的pbs-H1S质粒本课题构建,pbluescript II(KS+)由本实验室保存。
1.1.2 主要培养基及试剂①G.hansenii
ATCC23769用HS培养基:葡萄糖2%,蛋白胨0.5%,酵母浸出物0.5%,柠檬酸0.114%,Na2HPO4·12H2O 0.68 g/L;②大肠埃希菌培养用LB培养基(筛选平板含100 μg/mL Amp);③电泳用TBE电泳缓冲液;④淀粉水解酶活定性分析采用淀粉-琼脂鉴定平板(可溶性淀粉0.025 g,琼脂2 g,加dH2O定容至100 mL,121℃灭菌20 min)和卢戈氏(Lugol's)碘液;⑤测定淀粉水解酶活性用DNS试剂。
1.2 方法
1.2.1 黑曲霉RNA的提取和cDNA反转录黑曲霉总RNA提取:将黑曲霉保藏菌种经查氏斜面培养基活化后,接种于100 mL灭菌的黑曲霉液体发酵培养基中,230 r/min、30℃,培养24 h。离心收集黑曲霉菌丝体,抽滤去除菌丝体的水分,加入预冷的研钵中,加液氮充分研磨后,将粉末加到1 mL Trizol提取缓冲液中,提取步骤参照Trizol产品说明书。1.0%的琼脂糖测扩增凝胶电泳检产物。利用oligo(dT)12-18Primer进行反转录,具体步骤依照产品说明书。
1.2.2 胞外分泌的淀粉表达载体plac-amyE、placUV5-amyE的构建本实验使用的所有引物序列如表1。

表1 引物序列Table 1 The primer sequence
以pUC18质粒为模板,设计Lac-F和Lac-R引物扩增得到lac启动子,为消除葡萄糖效应,提高启动效率,进一步设计了LacUV5-R引物,与引物Lac-F一起,扩增得到不受葡萄糖抑制的Lac-UV5启动子。引物Lac-F、Lac-R和LacUV5-R的序列见表1。以G.hansenii ATCC23769基因组DNA为模板,设计sig-F和sig-R引物,扩增ATCC23769的胞外蛋白内-β-1,4-葡聚糖酶(endo-β-1,4-glucanase,CMCax;EC 3.2.1.4)信号肽片段SigP(该片段包含该菌自身的RBS位点和分泌胞外的信号肽,约180 bp)[6];以枯草芽胞杆菌Bacillus subtilis WB600基因组为模板,设计amyEF和amyE-R引物,扩增B.subtilis WB600的淀粉酶成熟肽(约1 900 bp)。将ApaⅠ-HindⅢ双切的pbs-H1S质粒与ApaⅠ-XbaⅠ双切的lac启动子/lacUV5启动子片段、XbaⅠ-BamHⅠ双切的sigP片段、BamHⅠ-HindⅢ双切的amyE片段同时连接,转化E.coli DH5α感受态细胞得到重组质粒(最终连接产物为lac/lacUV5启动子-sigP-amyE)。根据构建表达载体的内部酶切位点,选择BglⅡ对重组质粒进行单酶双切验证,琼脂糖凝胶电泳检测。
1.2.3 胞外分泌的糖化酶表达载体plac-GA的构建以G.hansenii ATCC23769基因组为模板,设计sig2-F和sig2-R引物,PCR扩增得到的信号肽片段Sig2(含该细菌自身RBS位点,约180 bp)。以黑曲霉A.niger cDNA为模板,设计Gla-F和Gla-R引物,扩增得到糖化酶基因片段(约1 900 bp)。将HindⅢ单酶切后去磷酸化的pbs-H1S质粒与HindⅢ-XbaⅠ双切sig2片段XbaⅠ-HindⅢ双切的糖化酶片段同时连接,转化E.coli DH5α感受态细胞得到重组质粒(最终连接产物为sig2片段-GA,启动子为pBS上自带的lac启动子)。根据构建表达载体的内部酶切位点,选择BamHⅠ对重组质粒进行单酶双切验证,琼脂糖凝胶电泳检测。
1.2.4 同时表达淀粉酶和糖化酶的表达载体plac-GA-amyE的构建采用Lac-newF和amyE-R引物,以质粒plac-amyE为模板,PCR扩增得到含lac-sigP-amyE片段(约2 300 bp)。采用Sig2-newF和Gla-newR引物,以质粒plac-GA为模板扩增得到sig2-GA基因片段(约2 000 bp)。将XhoⅠ-HindⅢ双切的pbs-H1S质粒与NotⅠ-HindⅢ双切的lac-sig-amyE片段、XhoⅠ-NotⅠ双切的sig2-GA片段同时连接,转化E.coli DH5α感受态细胞得到重组质粒(最终连接产物为lac启动子-sig2-GA-lac启动子-sigP-amyE)。根据构建表达载体的内部酶切位点,选择SalⅠ对重组质粒进行单酶双切验证,琼脂糖凝胶电泳检测。
1.2.5 表达amyE、GA和GA-amyE的葡糖酸醋杆菌的构建选取1~2 μL经过限制性酶切鉴定大小正确的plac-amyE、placUV5-amyE、plac-GA、plac-GA-amyE重组质粒,选取pUC18质粒做阴性对照,pbs-H1S作为阳性对照,电击转化进G.hansenii ATCC23769自发不产膜突变体(cel-)感受态菌。电转参数如下:0.1 cm电转杯(Bio-Rad),1.8 kV,200 Ω,25 μF。
1.2.6 淀粉平板透明圈检测酶活用枪头刮下平板上阳性克隆菌苔于6 mL HS(Amp 200 μg/mL)培养液中,28℃230 r/min,摇床培养过夜。转接(1/10)培养12 h,取培养后的菌液离心,取60 μL的发酵液上清,以原始G.hansenii ATCC23769 cel-菌液上清做阴性对照,枯草芽胞杆菌B.subtilis WB600菌液上清做阳性对照,37℃温育24 h后滴入少量的Lugol's碘液,进行淀粉平板透明圈检测。
1.2.7 DNS方法测定淀粉水解酶酶活根据3,5-二硝基水杨酸法(DNS法)[7],向15 mL具塞试管中加入1.2 mL 2%的淀粉溶液,于40℃水浴下保温3~5 min,然后加入酶液0.8 mL反应20 min,加入DNS试剂3 mL,混匀,沸水浴10 min,立即冷却定容至15 mL,540 nm测定吸光值。空白以煮沸失活的酶液代替。活力定义为:在本测定条件下,1 mL酶液在40℃条件下1 h水解2%淀粉产生1 mg葡萄糖所需的酶量定义为1个酶活力单位(U)。
2 结果与分析
2.1 plac-amyE和plac UV5-amyE外分泌穿梭表达载体的构建
用Lac-F和Lac-R引物以pUC18质粒为模板获得lac启动子,用Lac-F和lacUV5-R引物以pUC18质粒为模板PCR获得lacUV5启动子(图1)。

图1 lac启动子及lacUV5启动子的克隆Fig.1 The cloning of lac promoter and lacUV5 promoter
用SigP-F和SigP-R引物,以G.hansenii ATCC23769基因组为模板,获得该细菌自身的胞外蛋白内-β-1,4-葡聚糖酶(CMCax)的信号肽片段SigP(图2)。以枯草芽胞杆菌B.subtilis WB600基因组为模板,设计amyE-F和amyE-R引物,扩增淀粉酶基因amyE片段(图2)。

图2 SigP片段和amyE片段的克隆Fig.2 The cloning of SigP fragment and amyE fragment
对上述得到的亚克隆片段进行酶切,将酶切产物lac启动子/lacUV5启动子(ApaⅠ-XbaⅠ)、SigP(XabⅠ-BamHⅠ)及amyE(BamHⅠ-HindⅡ)与酶切的pbs-H1S(ApaⅠ-HindⅢ)质粒进行连接后,转化大肠埃希菌E.coli DH5α得到转化子,菌落PCR验证阳性克隆子,选取阳性克隆培养,提取质粒,利用重组质粒上的BglⅡ单酶双切鉴定(图3)。
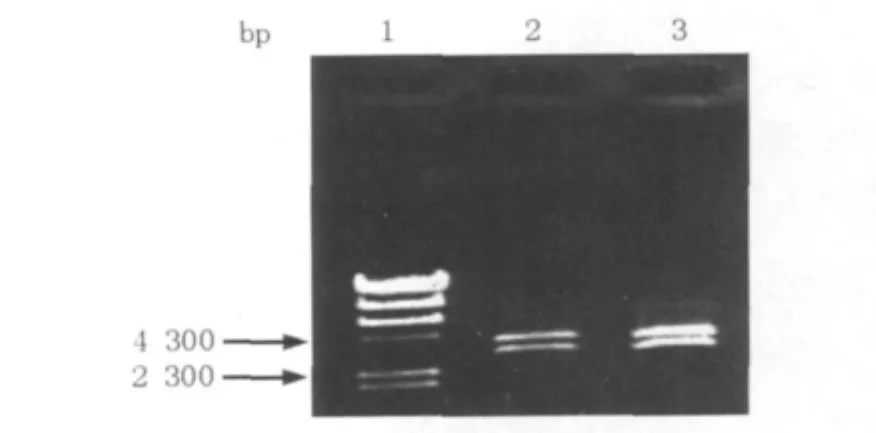
图3 重组质粒plac-amyE和placUV5-amyE的BglⅡ酶切鉴定Fig.3 Recombinant plasmid plac-amyE and placUV5-amyE digested by BglⅡ
电泳结果均出现了预期的大小3 084 bp和3 700 bp,说明重组质粒plac-amyE和placUV5-amyE构建成功。选取经过酶切验证正确的重组质粒plac-amyE和placUV5-amyE,电转化G.hansenii ATCC23769 cel-,28℃培养3 d。挑取长出的单克隆用lac-F和amyE-R引物进行菌落PCR验证(图4)。

图4 菌落PCR验证转化子Fig.4 Identification of the transformant by colony PCR
从图4可以看出,G.hansenii ATCC23769 cel-(plac-amyE)和G.hansenii ATCC23769 cel-(placUV5-amyE)均出现阳性克隆大小约2 000 bp的条带,说明plac-amyE和placUV5-amyE电转化G.hansenii ATCC23769成功。
2.2 plac-GA外分泌穿梭表达载体的构建
用Sig2-F和Sig2-R引物,以G.hansenii ATCC23769基因组为模板,获得CMCax的信号肽片段Sig2(图5)。以黑曲霉Aspergillus niger cDNA为模板,设计Gla-F和Gla-R引物,扩增糖化酶基因片段(图6)。

图5 Sig2片段的克隆Fig.5 The cloning of Sig2 fragment
对上述得到的克隆片段进行酶切,将酶切产物Sig2(HindⅢ-XabⅠ)、糖化酶(XabⅠ-HindⅢ)与酶切并去磷酸化的pbs-H1S(HindⅢ)质粒进行连接后,转化大肠埃希菌E.coli DH5α得到转化子,菌落PCR验证阳性转化子,选取阳性克隆培养,提取质粒,BamHⅠ单酶双切鉴定(图7)。电泳结果显示均出现了预期的大小2 000 bp和4 500 bp,说明重组质粒plac-GA构建成功。
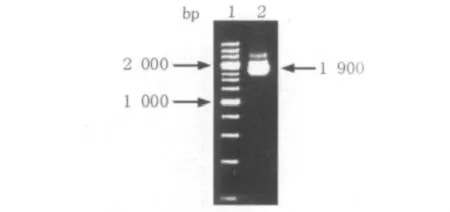
图6 糖化酶基因片段的克隆Fig.6 The cloning of glucoamylase fragment
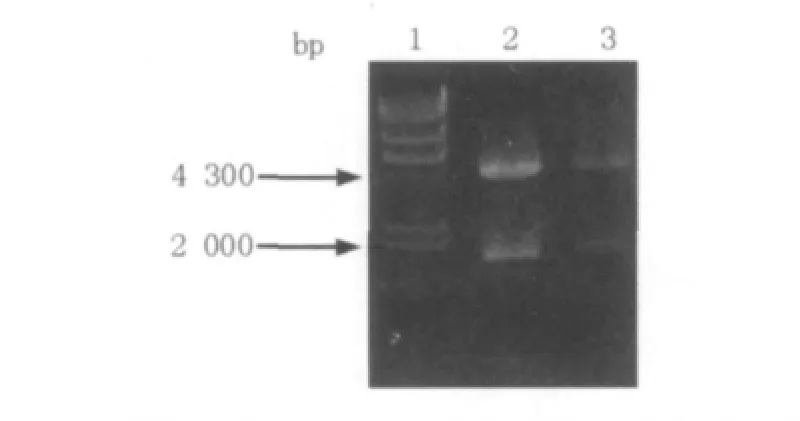
图7 重组质粒plac-GA的BamHⅠ酶切鉴定Fig.7 Recombinant plasmid plac-GA digested by BamHⅠ
选取经过酶切验证正确的重组质粒plac-GA,电转化G.hansenii ATCC23769 cel-,28℃培养3 d,挑取长出的单克隆用Gla-F和Gla-R引物进行菌落PCR验证(图8)。

图8 菌落PCR验证转化子Fig.8 Identification of the transformants by colony PCR
从图8可以看出,G.hansenii ATCC23769 cel-(plac-GA)出现阳性克隆大小约1 900 bp的条带,说明质粒plac-GA电转化G.hansenii ATCC23769成功。
2.3 plac-GA-amyE外分泌穿梭表达载体的构建
为了进一步在G.hansenii中同时表达淀粉酶和糖化酶,利用Sig2-newF和Gla-newR引物,以构建好的质粒plac-GA为模板,获得sig2-GA基因片段(图9)。利用Lac-newF和amyE-R引物,以质粒plac-amyE为模板,获得lac-sigP-amyE基因片段(图9)。
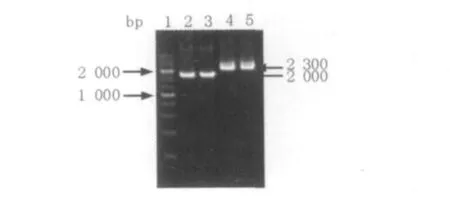
图9 sig2-GA片段及lac-sigP-amyE片段的克隆Fig.9 The cloning of sig2-GA fragment and lac-sigP-amyE fragment
对上述得到的片段进行酶切,将酶切产物sig2-GA(XhoⅠ-NotⅠ)、lac-sigP-amyE(NotⅠ-HindⅢ)与酶切的pbs-H1S(XhoⅠ-HindⅢ)质粒进行连接后,转化大肠埃希菌E.coli DH5α得到转化子,菌落PCR验证阳性克隆子,提取阳性克隆质粒,SalⅠ单酶双切鉴定,电泳显示预期的大小1 200 bp和7 500 bp,说明重组质粒plac-GA-amyE构建成功(图10)。在plac-GA-amyE中,糖化酶基因由pbluescript质粒上自身携带的lac启动子驱动,而淀粉酶基因由置于淀粉酶融合蛋白前的lac启动子驱动。
选取经过酶切验证正确的重组质粒plac-GA-amyE,电转化G.hansenii ATCC23769 cel-,28℃培养3 d,挑取长出的单克隆用Gla-F和Gla-R引物进行菌落PCR验证(图11)。
从图11可以看出,G.hansenii ATCC23769 cel-(plac-GA-amyE)出现阳性克隆大小约1 900 bp的条带,说明plac-GA-amyE电转化G.hansenii ATCC23769成功。

图10 重组质粒plac-GA-amyE的SalⅠ酶切鉴定Fig.10 Recombinant plasmid plac-GA-amyE digested by SalⅠ
2.4 验证表达淀粉酶和糖化酶的工程菌
选取上述验证为阳性的重组G.hansenii ATCC23769 cel-工程菌,用枪头刮下平板上阳性克隆菌苔于6 mL HS(Amp 200 μg/mL)培养液中,28℃230 r/min,摇床培养过夜。转接(1/10)培养12 h,取培养后的菌液离心,取上清,以原始G.hansenii ATCC23769 cel-菌液上清做阴性对照,枯草芽胞杆菌B.subtilis WB600菌液上清做阳性对照,进行淀粉平板透明圈检测(图12)。从图12中可以看出,G.hansenii ATCC23769 cel-周围没有水解圈,说明其本身不能表达淀粉酶。而转入plac-amyE、placUV5-amyE、placGA及

图11 菌落PCR验证转化子Fig.11 Identification of the transformants by colony PCR
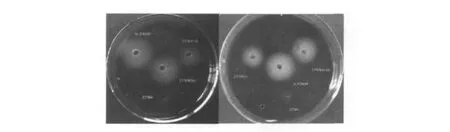
图12 碘-淀粉平板透明圈检测Fig.12 Iodine-starch flat hydrolysis circle test
plac-GA-amyE载体的ATCC23769工程菌,其周围都出现了明显的淀粉水解圈,说明构建的一系列工程菌都能表达具有淀粉水解活性的酶,且融合蛋白都能正常的分泌到细胞之外。
2.5 DNS方法测淀粉酶酶活
用DNS方法测得酶活力(表2)。

表2 淀粉水解酶的酶活力Table 2 Amylolytic activities
DNS结果显示,各种表达载体在葡糖酸醋杆菌中都能成功表达且分泌到细胞之外,但是所获得的酶活偏低。placUV5-amyE与plac-amyE相比,虽然采用了不受葡萄糖效应影响的lacUV5启动子,但是酶活反而略有下降,可能是由于采用lacUV5使得该基因表达不受调控导致细菌的生长受到影响,从而表现出酶活下降。因此后续表达糖化酶和糖化酶-淀粉酶的载体,只采用正常的lac启动子。较低的酶活结果说明lac启动子虽然能在葡糖酸醋杆菌中驱动外源基因的表达,但是其启动子效率偏低,无法有效水解淀粉以供葡糖酸醋杆菌生长和产细菌纤维素之用,因此需要寻找其他更有效的启动子。
3 讨论
细菌纤维素具有植物纤维素无法比拟的优点,因而具有广阔的运用前景。但细菌纤维素生产成本过高,直接影响其进一步工业化应用。针对其纤维素合成代谢途径的改造,力图降低细菌纤维素的生产成本成为很多人研究的目标。利用自主复制质粒在G.xylinus中表达绿豆的蔗糖合酶使细菌能利用蔗糖合成纤维素[8],以及在G.xylinus基因组上整合E.coli lacZ基因,使细菌能够利用廉价的乳清中的乳糖[9]。本研究利用能在G.hansenii自主复制的载体,在G.hansenii ATCC23769内源表达B.subtilis WB600的淀粉酶基因和A.niger的糖化酶GA基因,并进一步共表达淀粉酶基因和糖化酶基因,使G.hansenii具有了淀粉酶活性和糖化酶活性。经过淀粉平板透明圈检测,与B.subtilis WB600的阳性对照和G.hansenii ATCC23769 cel-的阴性对照,证明本实验所构建的工程菌都能成功表达有活性的淀粉水解酶。遗憾的是,利用DNS的定量检测显示构建的基因工程菌表达的淀粉酶和糖化酶的活性过低,无法满足降解淀粉供细菌生长及合成纤维素之需要。先前的研究有报道利用lac启动子来驱动外源基因在木葡萄酸醋杆菌中表达,如在G.xylinus中表达绿豆的蔗糖合酶使其能够利用蔗糖合成纤维素[8],但在本次试验中所取得的效果并不理想。其原因可能是lac启动子在该细菌中的启动子强度不高,或者由于CMCax的信号肽并不足以有效地将表达的外源蛋白分泌到胞外导致有活性蛋白含量少。因此换用其他的启动子进行验证,如组成型的β-内酰胺酶启动子(bla启动子)也曾被用来在G.xylinus中成功表达外源基因[10-12],或者寻找葡萄酸醋杆菌内源高效的启动子以及验证外源蛋白的分泌能力是下一步研究的目标。
[1] Shoda M,Sugano Y.Recent advances in bacterial cellulose production[J].Biotechnol Bioprocess Eng,2005,10(1):1-8.
[2] Putra A,Kakugo A,Furukawa H,et al.Tubular bacterial cellulose gel with oriented fibrils on the curved surface[J].Polymer,2008,49(7):1885-1891.
[3] Ross P,Mayer R,Benziman M.Cellulose biosynthesis and function in bacteria[J].Microbiol Rev,1991,55(1):35-58.
[4] Iyer PR,Geib SM,Catchmark J,et al.Genome sequence of a cellulose-producing bacterium,Gluconacetobacter hansenii ATCC 23769[J].J Bacteriol,2010,192(16):4256-4257.
[5] Ramana K,Tomar A,Singh L.Effect of various carbon and nitrogen sources on cellulose synthesis by Acetobacter xylinum[J].World Journal of Microbiology and Biotechnology,2000,16(3):245-248.
[6] Kawano S,Tajima K,Kono H,et al.Effects of endogenous endo-beta-1,4-glucanase on cellulose biosynthesis in Acetobacter xylinum ATCC23769[J].J Biosci Bioeng,2002,94(3):275-281.
[7] Miller GL.Use of dinitrosalicylic acid reagent for determination of reducing sugar[J].Analytical chemistry,1959,31(3):426-428.
[8] Nakai T,Tonouchi N,Konishi T,et al.Enhancement of cellulose production by expression of sucrose synthase in Acetobacter xylinum[J].Proc Natl Acad Sci U S A,1999,96(1):14-18.
[9] Battad-Bernardo E,McCrindle SL,Couperwhite I,et al.Insertion of an E.coli lacZ gene in Acetobacter xylinus for the production of cellulose in whey[J].FEMS Microbiol Lett,2004,231(2):253-260.
[10] Chien LJ,Chen HT,Yang PF,et al.Enhancement of cellulose pellicle production by constitutively expressing vitreoscilla hemoglobin in Acetobacter xylinum[J].Biotechnol Prog,2006,22(6):1598-1603.
[11] Yadav V,Paniliatis BJ,Shi H,et al.Novel in vivo-degradable cellulose-chitin copolymer from metabolically engineered Gluconacetobacter xylinus[J].Appl Environ Microbiol,2010,76(18):6257-6265.
[12] Setyawati MI,Chien LJ,Lee CK.Expressing Vitreoscilla hemoglobin in statically cultured Acetobacter xylinum with reduced O(2)tension maximizes bacterial cellulose pellicle production[J].J Biotechnol,2007,132(1):38-43.
猜你喜欢
纺织科技进展(2021年3期)2021-06-09
陶瓷学报(2021年1期)2021-04-13
食品与生物技术学报(2021年5期)2021-01-16
中国粮油学报(2019年4期)2019-07-12
中国麻业科学(2019年2期)2019-06-18
中国酿造(2019年4期)2019-05-09
食品与生物技术学报(2019年12期)2019-02-15
天然产物研究与开发(2018年6期)2018-07-09
应用化工(2014年11期)2014-08-16
中国酿造(2014年9期)2014-03-11
