
2-DG增强乳腺癌细胞对阿霉素化疗敏感性的作用
2010-11-29 09:23:26宋乐乐马琳艳蒋国君张旭东蒋志文
中国药理学通报 2010年10期
程 秀,刘 浩,方 琳,苏 方,宋乐乐,马琳艳,蒋国君,张旭东,蒋志文
乳腺癌是一种严重危害女性身心健康的恶性肿瘤,发病率在全球范围内位居女性恶性肿瘤的首位,并以每年2%的速度递增。全球每年新增病例120万,死亡病例50万,据估计到2010年全球每年新增病例将达150万人[1]。以手术为主,配合化疗、放疗、内分泌疗法、免疫疗法及中医药疗法的综合治疗措施,是目前治疗乳腺癌高效低毒的优化方案[1-2]。但是肿瘤对大多数化疗药耐药,对化疗药引起的细胞凋亡不敏感,因此,研究其耐药机制、克服化疗耐药,发现新的治疗靶点是肿瘤研究的热点之一[3-4]。本研究观察了糖基化抑制剂2-DG对乳腺癌细胞增殖及凋亡的影响,并联合临床常用治疗乳腺癌的药物阿霉素,检测2-DG增强ADM诱导乳腺癌细胞凋亡的作用,并对其初步机制进行探讨,以期为乳腺癌的临床治疗提供新的思路和方案。
1 材料与方法
1.1 材料
1.1.1 细胞 细胞系Sk-Br-3(购于美国ATCC公司),由蚌埠医学院生化药理研究室冻存培养。
1.1.2 试剂 DMEM高糖培养基、胰蛋白酶、Hepes、新生牛血清:Gibco公司。2-DG:Sigma公司;阿霉素:浙江海正药业股份有限公司;Caspase-3检测试剂盒:碧云天生物技术研究所。
1.2 方法
1.2.1 细胞培养 乳腺癌肿瘤细胞采用DMEM高糖培养液,10%(V/V)FBS,100 U·ml-1青霉素,100 mg·L-1链霉素,37℃、5%CO2饱和湿度下培养。
1.2.2 MTT 将细胞按每孔200 μl(含1×104个细胞)种于96孔板,每5孔为一组,每种药取5个质量浓度,各组药物终浓度如下,ADM:0.625、1.25、2.5、5、10 mg·L-1。另设阴性对照组(不加药物)和空白对照组(不加细胞,只加培养基)。终止培养前4 h,每孔加MTT 20μl(5 g·L-1),加药后24~72 h(药物作用终点时间)终止培养,弃去上清液,加入二甲亚砜200 μl每孔,微量振荡器摇震5 min,全自动定量绘图酶标仪测定每孔的595 nm波长吸光度A值,各药重复试验3组。
1.2.3 溴化丙啶(propidium iodide,PI)染色检测细胞凋亡 将对数生长期细胞制成单细胞悬液接种于24孔细胞培养板,每孔1×105个细胞,培养16~24 h后按实验设计加入药物,继续培养48h收集各孔培养液至对应流式管中,用预冷的PBS 500 μl清洗培养孔,收集清洗液至对应培养液中,1 200 r·min-1,离心10 min,弃上清;培养板中加入PI缓冲液(5 mg PI,0.1 g 柠檬酸钠、100 μl TritonX-100、100 ml dH2O)750 μl/孔,37℃ 孵育 10 min,吹打收集细胞至对应流式管中,轻轻摇动混匀。4℃避光保存,过夜,上流式细胞仪检测。利用PI染色,检测具有亚G1期DNA含量的细胞比例,代表凋亡细胞数。
1.2.4 Caspase-3活性检测 按照说明书进行检测,反应温度为 37℃。取样本 100 μl,Ac-DEVDAMC(Caspase-3四肽荧光底物)10 μl加到HEPES缓冲液中,在37℃下作用1 h,应用荧光分光光度计在波长405 nm处测吸光度,以不加底物的样本作空白,结果以吸光度值表示。
1.2.5 Western blot检测蛋白表达 收集稳定表达细胞用细胞裂解液(总体积200 ml:100 mmol·L-1Tris-HCl pH 7.4 20 ml、1 mol·L-1NaCl 28 ml、100 mmol·L-1CaCl21 ml、100 mmol·L-1MgCl221 ml、15 mmol·L-1NaN340 ml、Triton X-100 2ml,用前加入蛋白酶抑制剂 12 μmol·L-1Leupeptin、1 mmol·L-1PMSF各2 ml·L-1)冰上裂解30 min,提取细胞总蛋白,BCA蛋白定量法(参照试剂盒说明书操作)测各组蛋白浓度,用细胞裂解液将各组蛋白稀释至等浓度,与2×上样缓冲液1∶1混合,100℃煮沸5 min蛋白变性。取蛋白 30 μg/组,10%SDS-PAGE凝胶电泳(70 V,30 min;150 V,90 min);转膜(50 V,90 min)至硝酸纤维素膜;5%脱脂牛奶室温封闭2 h(或4℃过夜);一抗:1∶250,室温孵育2 h(或4℃过夜);TPBS洗涤3次;二抗:1∶2500,室温孵育2 h;TPBS洗涤3次,PBS洗涤1次;ECL发光试剂盒暗室发光、显影、定影。Bio-Rad凝胶成像系统获取图像。
1.2.6 集落克隆实验 用含体积分数10%FBS的培养基调整细胞浓度后,分别以每孔800个细胞接种于6孔板中,24 h后加0.132 mg·L-1ADM,1 mmol·L-12-DG以及两者合用处理乳腺癌细胞,置CO2孵箱中培养d 5,观察到细胞集落形成后,去除培养基,PBS洗涤2遍,20%甲醇 -20℃固定10 min,结晶紫染色,集落形成率的降低证实,2-DG增加ADM对乳腺癌细胞Sk-Br-3的增殖抑制作用。
2 结果
2.1 2-DG增强ADM对乳腺癌细胞Sk-Br-3增殖的抑制作用 为观察2-DG抑制乳腺癌细胞Sk-Br-3增殖的作用及对ADM抑制乳腺癌细胞增殖作用的影响,实验中使用不同浓度的2-DG刺激细胞,并联合ADM,使用MTT法检测细胞存活率。结果表明,10 mmol·L-12-DG作用于乳腺癌细胞Sk-Br-3的24、48、72 h 的存活率分别为 80.73%、57.16%、70.10%。ADM作用于乳腺癌细胞Sk-Br-3的24、48、72 h增殖抑制率分别为 76.89%、76.79%、70.07%。10 mmol·L-12-DG对 0.625 mg·L-1ADM诱导乳腺癌细胞Sk-Br-3 24、48、72 h的存活率分别为67.18%、67.52%、67.13%。ADM、2-DG与2-DG诱导ADM乳腺癌细胞增殖抑制率差异有统计学意义(P<0.05),见Fig 1。
2.2 2-DG增强ADM诱导乳腺癌细胞Sk-Br-3凋亡的作用 实验中对不同因素处理的细胞进行PI染色,并通过流式细胞仪分析,观察药物诱导乳腺癌细胞凋亡的作用,结果见Fig 2。10 mmol·L-12-DG刺激48h,诱导乳腺癌细胞 Sk-Br-3的凋亡率为5.8%(Fig 2B),与阴性对照组0.16%比较差异无统计学意义(P>0.05,Fig 2A)。0.4 mg·L-1ADM 诱导乳腺癌细胞Sk-Br-3 48 h的凋亡率为35.12%(Fig 2C)。10 mmol·L-12-DG 对 0.4 mg·L-1ADM诱导乳腺癌细胞Sk-Br-3 48 h的凋亡率为58.11%(Fig 2D),与2A及2B比较差异有统计学意义(P<0.05)。上述表明,2-DG本身并不诱导细胞凋亡,但可以增强阿霉素诱导的乳腺癌细胞凋亡。
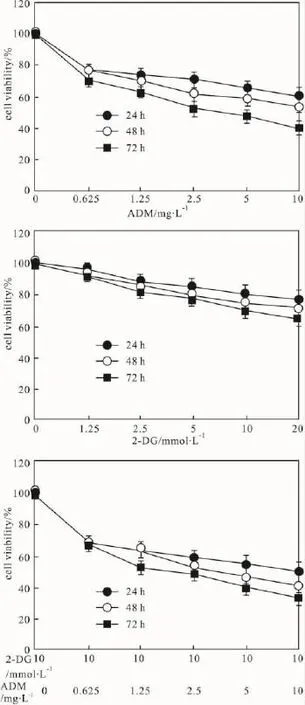
Fig 1 MTT conversion after treated with 2-DG,ADM and 2-DG combination with ADMSk-Br-3 cells were incubated with the indicated concentrations of 2-DG,ADM and 2-DG combination with ADM for 24 h,48 h and 72 h.MTT was added the amount of reduced formazan product determined spectrophotometrically.Results are expressed as percent of control non-treated cells.
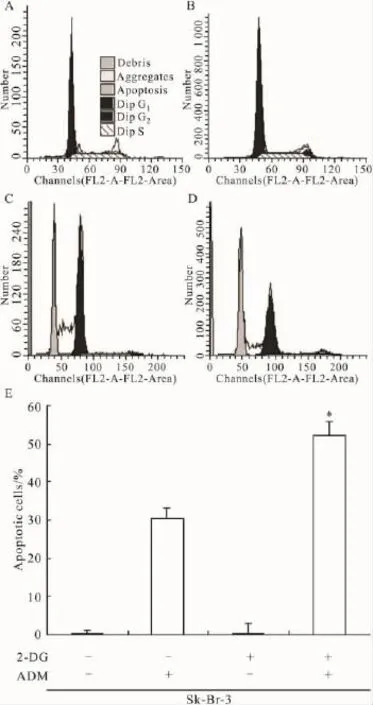
Fig 2 ADM-induced Sk-Br-3 apoptosis was increased by 2-DGA,B and C show that Sk-Br-3 cells were treated with 2-DG(10 mmol·L-1),ADM(0.4 mg·L-1)and 2-DG+ADM for 48 h.Sub-G1 populations among the Sk-Br-3 cells were counted by FACS,and the data are expressed as the ¯x± s for the three determinations in triplicate(*P <0.05 vs control).
2.3 2-DG增强ADM对乳腺癌细胞Sk-Br-3的集落克隆形成的抑制作用 为进一步观察2-DG抑制乳腺癌细胞Sk-Br-3增殖的作用及对ADM抑制乳腺癌细胞Sk-Br-3增殖作用的影响,实验中使用1 mmol·L-12-DG刺激细胞,并联合0.1 mg·L-1ADM,使用集落克隆形成实验检测乳腺癌细胞Sk-Br-3增殖作用的影响。Fig 3中,A、B、C、D分别是空白对照组、1 mmol·L-12-DG、0.1 mg·L-1ADM和1 mmol·L-12-DG+0.1 mg·L-1ADM处理Sk-Br-3细胞,给药处理5 d后,20%甲醇-20℃固定10 min,结晶紫染色。结果表明,2-DG在低浓度即可明显抑制Sk-Br-3细胞的集落克隆形成,并可增强ADM的抑制作用。

Fig 3 2-DG increased the colony formation inhibited effect of ADM on Sk-Br-3 cellsB,C and D:Sk-Br-3 cells were treated with 1 mmol·L-12-DG,0.1 mg·L-1ADM and 1 mmol·L-12-DG+0.1 mg·L-1ADM for five days.
2.4 2-DG增强ADM诱导乳腺癌Sk-Br-3细胞的Caspase-3激活作用 为观察2-DG诱导乳腺癌细胞Sk-Br-3的Caspase-3的活性改变及对ADM诱导乳腺癌细胞Sk-Br-3的Caspase-3的活性增加的影响,根据Caspase-3活性试剂盒检测Caspase-3活性,结果表明:2-DG刺激细胞后,并未出现Caspase-3的激活,但2-DG对ADM诱导乳腺癌细胞Sk-Br-3的Caspase-3激活有明显的增强作用(P<0.01)。
2.5 2-DG 对ADM诱导的Sk-Br-3的GRP-78及Caspase-3蛋白表达的影响 Sk-Br-3给予不同的刺激后收集蛋白,Western blot检测 GRP-78以及Caspase-3蛋白的表达,从Fig 5可以看出,2-DG可诱导GRP-78表达,随时间延长,GRP-78的表达不断增强。2-DG联合ADM处理Sk-Br-3细胞后,可诱导更为明显的GRP-78的表达。2-DG本身并未诱导Caspase-3蛋白的表达,联合ADM后激活型的Caspase-3蛋白表达明显增加。
3 讨论
糖基化抑制剂2-DG的抗肿瘤作用被广泛关注,从培养细胞到实验动物,以及临床实验不断的开展,并针对单独抗肿瘤到与其他抗肿瘤药物联合应用方面进行探索。同其他肿瘤一样,乳腺癌必须依赖于对葡萄糖摄取的增加来维持自身的快速增殖。本次研究观察了2-DG增强ADM抗乳腺癌的作用。从上述的结果可以看出,2-DG可增加ADM对乳腺癌细胞Sk-Br-3的增殖抑制作用,增强其诱导的肿瘤细胞凋亡;2-DG还可增高ADM诱导乳腺癌细胞Sk-Br-3的GRP-78表达,引起内质网应激反应,并增强Caspase-3蛋白的表达。
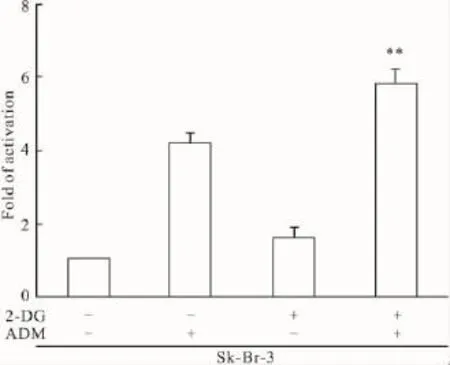
Fig 4 2-DG enhanced ADM induced activation of casepase-3 in Sk-Br-3Cells treated with 2-DG(10 mmol·L-1),ADM(0.4 mg·L-1),2-DG+ADM.Sk-Br-3 cells were treated with 10 mmol·L-12-DG,0.4 mg·L-1ADM,2-DG+ADM for 6 h.Cells were harvested and Caspase-3 activity measured using a spectrophotometric assay according to the manufacturer′s instructions.Control cells were incubated for 6 h in the presence of media only.**P<0.01 vs 2-DG
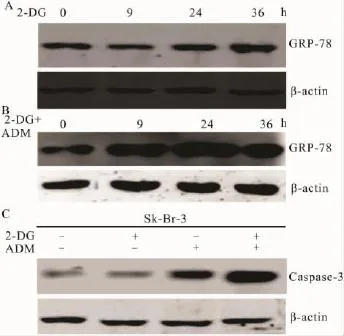
Fig 5 Expressions of GRP-78 and Caspase-3 in Sk-Br-3 cellsA:2-DG induced activation of UPR in Sk-Br-3 cells;B:2-DG+ADM induced activation of UPR in Sk-Br-3;C:2-DG activation Caspase-3 in Sk-Br-3 cells.
葡萄糖调节蛋白78(glucose-regulated protein 78,GRP-78)是内质网(endoplasmic reticulum,ER)功能的中心调节者,由于其在蛋白质折叠、聚集、促进非折叠蛋白降解、ER Ca2+连接和控制ER跨膜感受器的激活等过程中具有重要作用,因此GRP-78的诱导被广泛用作ER应激和UPR启动的生物标记[5-6]。体外研究显示[7],肿瘤细胞程度性地合成GRP-78维持内环境的稳定,构筑自身防御机制。在生长环境恶劣的肿瘤微环境中,乳腺癌细胞自身发生ER应激激活UPR。给予2-DG时可影响肿瘤细胞对葡萄糖的摄取,降低ATP的产生,抑制肿瘤细胞增殖。实验结果中发现,2-DG可诱导乳腺癌细胞的ER应激,GRP-78表达明显增强,启动剧烈的UPR。由于ER应激的保护作用,削弱了细胞凋亡反应的发生,因此乳腺癌细胞对能够激活UPR的化疗药物介导的细胞凋亡不敏感。GRP-78的高表达则是乳腺癌细胞对ER应激途径的化疗药耐药的原因之一。可见,2-DG上调 GRP-78的表达可能是Sk-Br-3细胞对2-DG诱导的细胞凋亡不敏感的原因之一。但必须强调的是,UPR本身是一种细胞保护反应,但过度的UPR最终启动细胞凋亡进程[8]。因此,触发肿瘤细胞产生过强的ER应激反应,促使UPR保护机制无法产生抗凋亡作用,而激活下游Caspase的活性,实现肿瘤细胞的凋亡。本研究中发现,2-DG本身诱导乳腺癌细胞产生明显的ER应激反应,使得乳腺癌细胞Sk-Br-3仅出现增殖的抑制,而不诱导明显的凋亡作用,ER应激反应可能发挥保护作用,当其与ADM联合应用时,出现乳腺癌细胞凋亡明显增加,UPR机制并没有保护肿瘤细胞免于凋亡。检测ER应激标记物的结果发现,2-DG与ADM共同作用下,触发了更为强烈的ER反应,通过检测凋亡通路中Caspase-3活性的改变,也说明了凋亡的增加。这些结果说明了过度的ER应激可促使UPR从细胞保护转变成促进凋亡的现象存在。当然,关于ER应激的不同程度对肿瘤细胞的存活的影响尚需更多的实验进行说明[9],这也是本研究需要进一步深入探讨的问题,而本次研究恰为深入认识ER应激提供了一定的实验基础。
2-DG与葡萄糖结构的差别在于由氢替代第2个碳的羟基。而且2-DG有选择性地积聚在肿瘤细胞代谢中,这个可能归咎于提高了肿瘤细胞内的己糖激酶或细胞内磷酸化活性以及减少细胞内的磷酸盐的水平[10-11]。葡萄糖类似物2-DG可竞争葡萄糖的转运,抑制其吸收代谢,并降低 ATP产生[11-12]。体外和体内的实验均证实了葡萄糖类似物能抑制肿瘤细胞的葡萄糖代谢[13]。2-DG已被广泛研究作为一种可能的靶向治疗剂来治疗肿瘤,或者用来增强其他抗肿瘤药物的治疗效果[14-15]。乳腺癌和其他肿瘤细胞一样,都依赖于增加葡萄糖摄取,以维持细胞的生长,表现为葡萄糖摄取率增加并且葡萄糖转运蛋白表达增强。最近的研究表明[11],干扰糖酵解的非代谢葡萄糖类似物2-DG能通过干预糖酵解导致细胞死亡。2-DG能否调高临床肿瘤治疗疗效的研究也在不断深入,例如,2-DG可通过提高细胞表面TRAIL-R2的表达,而增强TRAIL诱导的黑色素瘤细胞的凋亡[16];2-DG还具有协同放射治疗增强乳腺癌细胞死亡的作用。本实验中发现了2-DG对乳腺癌细胞的增殖具有抑制作用,在联合临床常用抗肿瘤药物ADM时,证明了其增强ADM诱导乳腺癌细胞凋亡的作用。
总之,本研究证实了糖基化抑制剂2-DG对乳腺癌细胞的增殖具有抑制作用,但其本身并不诱导细胞凋亡,但可明显增加临床常用治疗乳腺癌细胞的药物诱导乳腺癌细胞凋亡的作用。其机制可能是通过诱导细胞产生过度的ER应激反应,增强Caspase-3活性而发挥作用。本研究为糖基化抑制剂的临床应用提供了实验基础,也为增强临床常用化疗药物疗效提供了一个良好的策略。但本实验中仅选用了体外培养的细胞株进行观察,进一步的体内外药效及机制有待深入研究。
[1]李 俊,翟所迪.临床药物治疗学[M].北京:人民卫生出版社,2007:415.
[1]Li J,Zai S D.Applied therapeutics the clinical use of drugs[M].Beijing:People′s Medical Publishing House,2007:415.
[2]Kim J,Han J Y,Shaw B,et al.Gustafson D:The roles of social support and coping strategies in predicting breast cancer patients′emotional well-being:testing mediation and moderation models[M].J Health Psychol,15:543-52.
[3]刘 浩,蒋志文,童旭辉,张旭东.硫酸乙酰肝素蛋白聚糖对C3H小鼠乳腺癌移植瘤的抑制作用及其机制[J].中国药理学通报,2008,24(6):744-8.
[3]Liu H,Jiang Z W,Tong X H,Zhang X D.Inhibitory effects of heparan sulfate proteoglycan on mice transplanted tumors[J].Chin Pharmacol Bull,2008,24(6):744-8.
[4]Simons A L,Mattson D M,Dornfeld K,Spitz D R.Glucose deprivation-induced metabolic oxidative stress and cancer therapy[J].J Cancer Res Ther,2009,5(Suppl 1):S2-6.
[5]Healy S J,Gorman A M,Mousavi-Shafaei P,et al.Targeting the endoplasmic reticulum-stress response as an anticancer strategy[J].Eur J Pharmacol,2009,625:234-46.
[6]Jiang C C,Chen L H,Gillespie S,et al.Tunicamycin sensitizes hu-man melanoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by up-regulation of TRAIL-R2 via the unfolded protein response[J].Cancer Res,2007,67:5880-8.
[7]蒋志文,LeBourhis X,Hubert H.肿瘤细胞的进行性增殖和Bip/GRP78 的合成[J].中国药理学通报,2002,18(1):79-83.
[7]Jiang Z W,Le B X,Hubert H.Progressing growth of tumor cell and synthesis of Bip/GRP78[J].Chin Pharmacol Bull,2002,18(1):79-83.
[8]Jiang C C,Wroblewski D,Yang F,et al.Human melanoma cells under endoplasmic reticulum stress are more susceptible to apoptosis induced by the BH3 mimetic obatoclax[J].Neoplasia,2009,11:945-55.
[9]Jiang C C,Lucas K,Avery-Kiejda K A,et al.Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress[J].Cancer Res,2008,68:6708-17.
[10]杨 芬,刘 浩,张旭东,蒋志文.抑制MEK对内质网应激诱导乳腺癌细胞凋亡的增敏作用[J].中国药理学通报,2009,25(6):778-82.
[10]Yang F,Liu H,Zhang X D,Jiang Z W.Inhibition of MEK sensitizes human breast carcinoma cells to endopla sm ic reticulum pa thway′s apoptosis[J].Chin Pharmacol Bull,2009,25(6):778-82.
[11]Yamada H,Takahashi N,Tanno S,et al.A selective orexin-1 receptor antagonist,SB334867,blocks 2-DG-induced gastric acid secretion in rats[J].Neurosci Lett,2005,376:137-42.
[12]Prasanna V K,Venkataramana N K,Dwarakanath B S,Santhosh V.Differential responses of tumors and normal brain to the combined treatment of 2-DG and radiation in glioablastoma[J].J Cancer Res Ther,2009,5(Suppl 1):S44-7.
[13]Varshney R,Adhikari J S,Dwarakanath B S.Contribution of oxidative stress to radiosensitization by a combination of 2-DG and 6-AN in human cancer cell line[J].Indian J Exp Biol,2003,41:1384-91.
[14]Dwarakanath B S,Singh D,Banerji A K,et al.Clinical studies for improving radiotherapy with 2-deoxy-D-glucose:present status and future prospects[J].J Cancer Res Ther,2009,5(Suppl 1):S21-6.
[15]Farooque A,Afrin F,Adhikari J S,Dwarakanath B S.Protection of normal cells and tissues during radio-and chemosensitization of tumors by 2-deoxy-D-glucose[J].J Cancer Res Ther,2009,5(Suppl 1):S32-5.
[16]Liu H,Jiang C C,Lavis C J,et al.2-Deoxy-D-glucose enhances TRAIL-induced apoptosis in human melanoma cells through XBP-1-mediated up-regulation of TRAIL-R2[J].Mol Cancer,2009,8:122.
猜你喜欢
中老年保健(2022年6期)2022-08-19 01:41:48
数学物理学报(2021年4期)2021-08-30 08:27:48
新世纪智能(数学备考)(2020年10期)2021-01-04 00:37:50
中国甜菜糖业(2020年1期)2020-12-07 07:54:30
中国生殖健康(2019年2期)2019-08-23 08:11:42
中国生殖健康(2019年6期)2019-01-06 09:20:12
江苏卫生保健(2018年7期)2018-07-31 08:35:34
祝您健康(2018年5期)2018-05-16 17:10:16
中成药(2017年12期)2018-01-19 02:06:52
中国交通信息化(2017年8期)2017-06-06 07:16:47
