
Al-Sc金属间化合物的电子结构及稳定性和热力学性质
2010-09-29 01:20采用基于密度泛函理论的赝势平面波法计算AlSc的4个稳定相的电子结构分析其成键情况并采用基于第一性原理的热力学计算公式计算各相的生成焓结合能德拜温度体弹模量和自由能结果表明AlSc金属间化合物在费米面以下主要通过AlScd和AlScd杂化成键在费米面以上则以Scd成键为主随着Al在各相中所占比例的增大体系中共价性增大稳定性提高且AlSc与AlSc在低能级区成键电子较多结构较为稳定4个化合物中K时合金化形成能力最强的是AlSckJmol最弱的是AlS
中国有色金属学报 2010年5期
采用基于密度泛函理论的赝势平面波法,计算Al-Sc的4个稳定相的电子结构,分析其成键情况,并采用基于第一性原理的热力学计算公式,计算各相的生成焓、结合能、德拜温度、体弹模量和自由能。结果表明:Al-Sc金属间化合物在费米面以下,主要通过Al s−Sc d和Al p−Sc d杂化成键,在费米面以上则以Sc d成键为主;随着Al在各相中所占比例的增大,体系中共价性增大,稳定性提高,且AlSc与AlSc在低能级区成键电子较多,结构较为稳定。在 4个化合物中, K时合金化形成能力最强的是 AlSc(H=−5. kJ/mol),最弱的是AlSc(H=−5.9 kJ/mol)。自由能计算所得曲线表明:AlSc的结构稳定性最好,AlSc的最差;随着温度的升高,晶体稳定性都降低。所有热力学计算值与试验值吻合良好。
关键词:Al-Sc金属间化合物;电子结构;稳定性;热力学性能;自由能;密度泛函理论;赝势平面波法
中图分类号:TG 111; TG146.2 文献标志码:A
Al-Sc金属间化合物的电子结构及稳定性和热力学性质
摘 要:采用基于密度泛函理论的赝势平面波法,计算Al-Sc的4个稳定相的电子结构,分析其成键情况,并采用基于第一性原理的热力学计算公式,计算各相的生成焓、结合能、德拜温度、体弹模量和自由能。结果表明:Al-Sc金属间化合物在费米面以下,主要通过Al 3s−Sc 3d和Al 3p−Sc 3d杂化成键,在费米面以上则以Sc 3d成键为主;随着Al在各相中所占比例的增大,体系中共价性增大,稳定性提高,且Al2Sc与Al3Sc在低能级区成键电子较多,结构较为稳定。在 4个化合物中,0 K时合金化形成能力最强的是 Al2Sc(H0=−52.02 kJ/mol),最弱的是AlSc2(H0=−35.93 kJ/mol)。自由能计算所得曲线表明:Al2Sc的结构稳定性最好,AlSc2的最差;随着温度的升高,晶体稳定性都降低。所有热力学计算值与试验值吻合良好。
关键词:Al-Sc金属间化合物;电子结构;稳定性;热力学性能;自由能;密度泛函理论;赝势平面波法
中图分类号:TG 111; TG146.2文献标志码:A
李燕峰1,2,徐 慧1,2,张 彪1,章立刚2
(1. 中南大学 物理科学与技术学院,长沙 410083;2. 中南大学 材料科学与工程学院,长沙 410083)
Abstract:Based on the density functional theory (DFT), the plane-wave pseudopotential method was used to calculate the electronic structures of four stable phases for Al-Sc alloys. The bonding situation was analyzed, and the formation enthalpy, binding energy, Debye temperature, bulk modulus and free energy of each phase were calculated by the thermodynamic calculation formula based on the first-principle. The results show that, under the Fermi energy, the bonds of each phase are mainly mixed by Al 3s-Sc 3d and Al 3p-Sc 3d; over the Feimi energy, those bonds are mainly contributed by electrons of Sc 3d. As the Al content increases, the covalent and stability of the system increase, and Al2Sc and Al3Sc are the stable structures by the more bonding electrons in low-energy area. Among the four crystals, the alloy-forming ability of Al2Sc(H0=−52.02 kJ/mol) is the greatest, that of the AlSc2(H0=−35.93 kJ/mol) is the lowest. The free energy curves show that the crystal stability of Al2Sc is the highest, AlSc2is the most unstable; and the stabilities of the four crystals are all reduced with increasing temperature. All the calculations data agree well with the experimental data.Key words:Al-Sc intermetallics compounds; electronic structure; stability; thermodynamic properties; Gibbs free energy;density functional theory; plane-wave pseudo-potential method
Al-Sc合金作为一种高强度、耐高温、高可焊性和耐腐蚀等优点于一体的新型结构材料[1−5],广泛应用于航空航天和核能工程等领域。随着计算技术的进步,人们开始通过计算获取Al-Sc金属间化合物的电子结构和热力学等参数,以完善实验数据体系。
TAO 等[6−7]用投影缀加平面波(PAW, Projector augmented-wave)法计算Al2Sc的形成焓和体弹模量,并用德拜−格奈森模型计算了Al-Sc的德拜温度、膨胀系数和热容等热力学参数。LU和WANG[8]利用VASP程序计算了Li 2结构的Al3Sc的基态能量,得出0 K时的生成焓和状态方程,并用(Mean field potential,MFP)[9]方法估算振动对有限温度下Gibbs形成能的影响。ASTA等[10−11]研究了Al-Sc的形成熵、体弹模量及其与电子结构的关系,指出大的形成熵主要来源于最近的Al与Sc的强键合作用。CACCIAMANI等[12]计算了Al-Sc系部分化合物的形成焓等热力学参数,并实验测量了部分反应的相平衡数据。
然而,至今对Al-Sc合金中AlxScy金属间化合物稳定相的热力学参数及电子结构还缺乏完整和系统的研究,且已有的数据甚至存在冲突[8,12]。在此,鉴于时效硬化高温铝合金的重要作用和Al-Sc合金在基础研究中的意义,本文作者通过结构驰豫得到晶体几何结构,计算弹性模量和德拜温度,分析AlxScy金属间化合物的电子结构,将温度引入热力学公式系统计算各化合物相的生成焓、结合能及Gibbs自由能,并与试验数据进行对比。
1 参数与方法
1.1 计算及试验方法
计算采用基于密度泛函理论的赝势平面波法,在晶体结构驰豫时使用 BFGS(Broyden-Fcetcher-Goldfarb-Shanne)运算法则,选取广义梯度近似(General gradient approximation, GGA)的PW91(Perdew and Wang)[13]形式来处理交换关联能部分,采用Vanderbilt形式的超软赝势来描述离子实与价电子之间的相互作用。在倒易的k空间中,通过平面波截断能(Ecut)来控制计算精度。计算中Ecut设为400 eV,自洽计算精度设为1.0×10−5eV/atom,布里渊区K点设为6×6×6。
1.2 结构驰豫
结合Al-Sc二元合金相图[12],选取的AlxScy金属间化合物包括AlSc2、AlSc、Al2Sc和Al3Sc 4种相,其各自的空间群和晶格参数如表1所列,建立的晶胞模型如图1所示。对4种晶胞进行结构驰豫后的几何参数的计算值也列于表1。

图1 Al-Sc金属间化合物的晶体结构模型Fig.1 Crystal structures models of Al-Sc intermetallics compounds: (a) AlSc2; (b) AlSc; (c) Al2Sc; (d) Al3Sc

表1 Al-Sc金属间化合物的空间群和晶格参数Table 1 Space group and lattice parameters of Al-Sc intermetallics compounds
2 电子结构分析
对AlSc2、AlSc、Al2Sc和Al3Sc的总态密度(Total density of states, TDOS)和分波态密度(Partial density of states, PDOS)进行计算,结果如图2所示。其中:Al的电子构型为 1s22s2p63s2p1,Sc的电子构型为 1s22s2p63s2p6d14s2;同时,将费米面移至零点处,仅考虑费米面附近态密度变化情况(费米面由点划竖线标出)。
分析发现,AlSc2的态密度(见图2(a))在费米能级以下主要由Al的3s、3p电子和Sc的3d电子杂化而成。其中,−7.5~−4.0 eV区域为Al 3s-Sc 3d杂化,−4.0~0 eV区域为Al 3p-Sc 3d杂化;费米面以上则为Sc的3d电子成键,即主要由Sc 3d贡献。费米面处在赝能隙谷底右方(反键态),该处几乎没有Al参与成键,电子密度较大而费米能级两侧尖峰间距很小,表明Al—Sc弱且AlSc2的局域稳定性较差。值得注意的是,费米面附近能带展宽较其他几个相的窄,且在费米面以下−4 eV处打开一个类似窄带隙半导体的带隙,表明其电子局域程度非常大,甚至可以通过电场等方法将费米面移动到带隙中而使其具有半导体性质。

图2 Al-Sc金属间化合物的TDOS和PDOS图Fig.2 TDOS and PDOS diagrams of Al-Sc intermetallics compounds: (a) AlSc2; (b) AlSc; (c) Al2Sc; (d) Al3Sc
AlSc的总态密度和部分态密度如图2(b)所示,可以看出其成键在费米能级以下区域与 AlSc2类似:小于−4.0 eV区域的态密度主要由 Al 3s-Sc 3d杂化,−4.0~0 eV区域为Al 3p-Sc 3d杂化;而在费米能级以上区域除了 Sc自身成键外,则出现了较为强烈的Al 3p-Sc 3d的杂化键。费米面位于赝能隙谷底,其所在处电子密度较小,由此可知AlSc晶体比较稳定。从图2(c)可以发现,随着Al量在各相中所占比例的增大,体系中出现Al—Al共价键(−10~−5 eV),费米面以上则仍是Sc—Sc键和部分的Al—Sc杂化键,费米面位于赝能隙谷底,且费米面处电子密度很小,说明Al2Sc局域稳定性很高,且费米面两侧峰距较大,表明
Al—Sc作用较强。Al3Sc与Al2Sc成键非常类似,而费米面在赝能隙谷底稍靠反键峰侧,两侧尖峰间距(赝能隙)较大,体现很强的共价性。值得注意的是,其费米能级附近的能带展宽比Al2Sc明显,由16 eV(Al2Sc)增大为 20 eV(Al3Sc),表明其电子非局域(Non-local)程度增大,共价性和原子轨道扩展性增强。
从成键峰的峰值和位置来看,AlSc2成键峰值(单位为electron/eV)为7(−1 eV)和3(−4.8 eV),AlSc为1.7(−2 eV)和0.8(−5.5 eV),Al2Sc为3.4(−1.6 eV)、2.5(−4 eV)、1.7(−6.9 eV)和1.5(−8.4 eV),Al3Sc为2.9(−2.5 eV)、2.4(−1.5 eV)、1.6(−4.1 eV)、1.5(−5.3 eV)和1(−8.3 eV和−6.7 eV)。以上表明,AlSc2成键峰位于费米能级附近,活性较大,而Al2Sc和Al3Sc的成键峰位置相对其他两化合物向低能级区移动,且在低能级区Al2Sc成键电子数最多,Al3Sc次之。同时,不难发现,Al3Sc和Al2sc的赝能隙均比Al2Sc和AlSc的更宽。
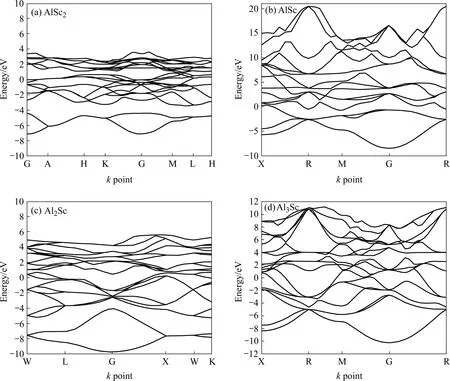
图3 Al-Sc金属间化合物的能带结构图Fig.3 Band structures of Al-Sc intermetallics compounds: (a) AlSc2; (b) AlSc; (c) Al2Sc; (d) Al3Sc
计算得到的4种化合物的能带结构如图3所示。从图3可以更加清晰地看到:4个化合物导带(即在高对称点附近近似成开口向上的抛物线形状的能带)均与费米能级相交并且多次穿越,表明各相均为金属性体系;但AlSc和Al3Sc在费米能级上、下两部分,即导带和价带处的能带均呈现明显的抛物线型,呈现类sp带特征,体现出较大的共价键杂化成分,即体系呈现较强的共价性。结合前面 DOS的赝能隙对比分析有,Al3Sc的赝能隙比AlSc的更宽,表明Al3Sc比AlSc更稳定。同时,从能带展宽来看,相对AlSc2,Al2Sc能带展宽较大,表明Al2Sc的非局域程度较大。结合前面DOS的成键峰分析可知,Al2Sc和Al3Sc体系费米能级以下较多的成键电子位于低能级区,表明两者在合金中是具有较高稳定性的相结构[20]。
综上分析表明,Al2Sc和Al3Sc在合金中具有较强的键合强度和共价性,其结构相对AlSc和AlSc2较为稳定,而AlSc2表现出较强的活性。
3 热力学公式及计算
3.1 生成焓与结合能的计算公式及结果
由热力学理论可知,对于Al-Sc金属间化合物,0 K时的生成焓H0为
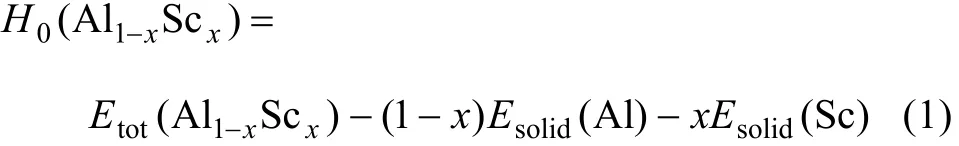
由晶体生成定义可知,Al-Sc化合物的结合能Ecoh为

式中:Etot(Al1−xScx)为Al1−xScx金属间化合物在平衡晶格常量下每个原子的平均总能量;Esolid(Al)和Eatom(Al)分别为Al在固态和自由状态(游离态)下平均每个原子的总能量;Esolid(Sc)和Eatom(Sc)分别为Sc在固态和自由状态(游离态)下平均每个原子的总能量;x为合金中Sc的摩尔分数。
计算得到的生成焓和结合能列于表2。从表2可以发现,AlSc2、AlSc和Al3Sc的H0计算值与文献吻合很好,误差在1 kJ/mol左右,只有Al2Sc与其他值存在较大偏差。由于生成焓的大小反映合金化的难易程度,Al2Sc生成焓最负,表明其合金化形成能力最强,即Al2Sc最易形成,Al3Sc次之,AlSc2最难形成且不稳定,这与关于态密度的分析结果一致。从结合能的数据也可看出,在0 K时稳定的是Al3Sc和Al2Sc,不稳定的是AlSc2。

表2 Al-Sc金属间化合物的生成焓和结合能Table 2 Formation enthalpy and cohesive energy of Al-Sc intermetallics compounds
为了获得高温时4种晶体的热稳定性,还分析计算Gibbs自由能随温度变化的情况。自由能的计算基于第一性原理和德拜理论,当忽略电子熵时体系的Gibbs自由能表达式[23]为)
(
(0TST
(T)
GT)=H
D
D-
+E(3)
式中:H0为0 K时的生成焓;ED和SD分别为温度为T时的振动能和振动熵。根据德拜理论,振动能和振动熵可用德拜温度表示:

式中:n为非负整数;θd为德拜温度。
将式(4)和(5)代入式(3),得到Gibbs自由能表达式如下:

同时,对于德拜温度有:

式中:ħ和kB分别为约化普朗克常数和玻尔兹曼常数;ωD为截止频率。
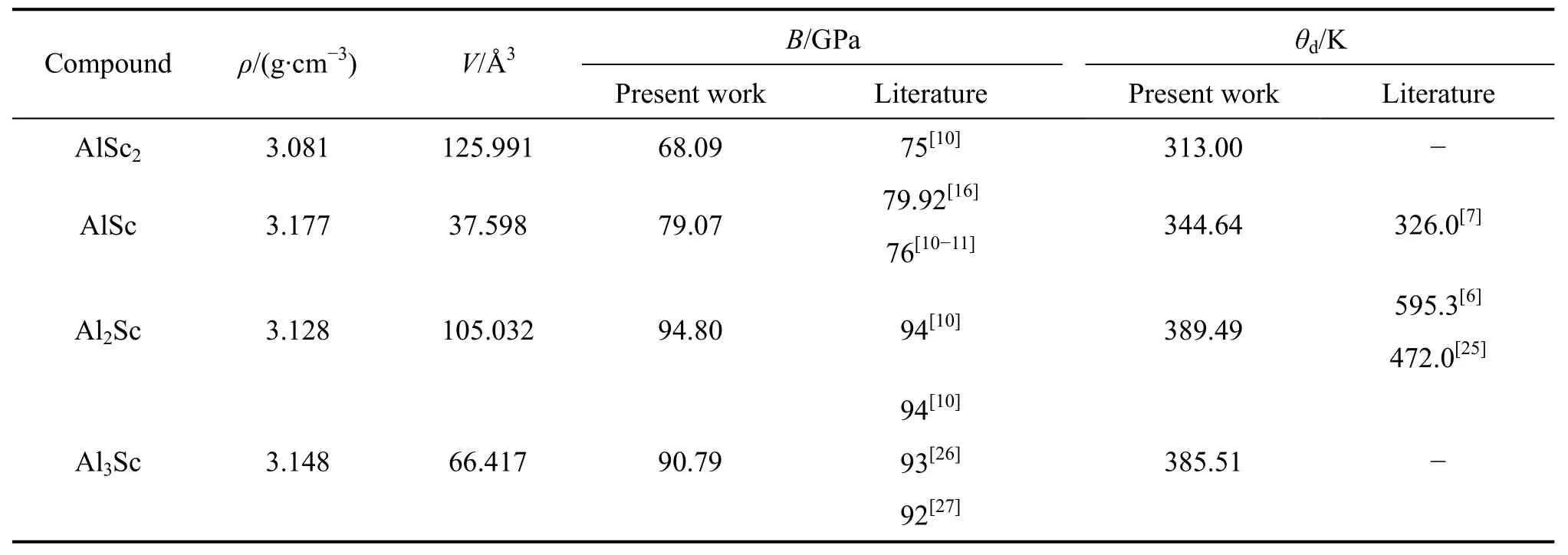
表3 Al-Sc金属间化合物的密度、体积、体弹模量和德拜温度Table 3 Densities, volumes, bulk modulus and Debye temperatures of Al-Sc intermetallics compounds

式中:V、N和vp分别为原胞体积、原胞原子个数和德拜平均声速。
MORUZZI等[24]研究发现vp可近似取
ρ
B
.617 0 。其中B为体弹模量;ρ为密度。则德拜温度可写为
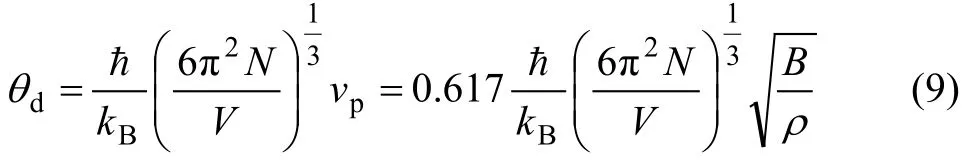
联合式(6)和(9),即可计算得到体弹模量、德拜温度,进而得到带温度参数的自由能G(T)。计算得到的中间参数,体弹模量B和德拜温度见表3所列。
由表3可以看出,体弹模量和德拜温度的计算数据与已有试验数据吻合较好,仅Al2Sc的德拜温度与文献[6]的有较大差别,这是因为文献[6]采用的晶格常数为7.573 Å,比本研究采用的优化弛豫(7.490 Å)大,且其采用的投影缀加波方法在晶格波函数的描述上为全电子方法,即对电荷密度进行进一步的分解,将波函数由平面波部分、赝波函数、每个原子和赝原子轨道的展开多次叠加而成,因而其结果比本研究的超软赝势方法和文献[25]的数值大。
采用上述值计算得到各化合物在100~1 500 K的摩尔自由能数值,如图4所示。
由图4可看出,4种晶体的Gibbs自由能都随温度升高而减小。由于高温时该4种化合物均已完全分解(Sc的熔点为1 539 ℃,Al的熔点为660 ℃),因此,未画出1 500 K以上的自由能曲线。从图4还可看到,晶格振动对自由能的贡献随着温度升高而慢慢减小各相之间的自由能差,但在已计算的温度范围内始终是Al2Sc的自由能最小,AlSc2最大,即随着温度的升高其结构稳定性都有所下降,但同一温度下始终是Al2Sc最稳定,AlSc2最不稳定。上述均符合有关前面态密度和能量计算分析的结论,并与文献[12]的结果一致。
值得注意的是,对于AlSc和Al3Sc在400 K左右时(见图4),自由能曲线相交,表明低温反应时 AlSc比Al3Sc更易生成。Sc在Al中的溶解度在655 ℃时为0.32%,在527 ℃时为0.07%,在室温下为零[28]。由此可见,溶解度随温度的降低而急剧降低。尽管低温反应时,AlSc比Al3Sc更易生成,但由于实际低温时(400 K),Sc的溶解度低造成Al富余,且通过前面DOS分析可知,Al3Sc成键更为稳定,因此,得到的均为密度很高的Al3Sc相。这可以用来解释试验观察到的情况与计算数据之间存在差别的原因。
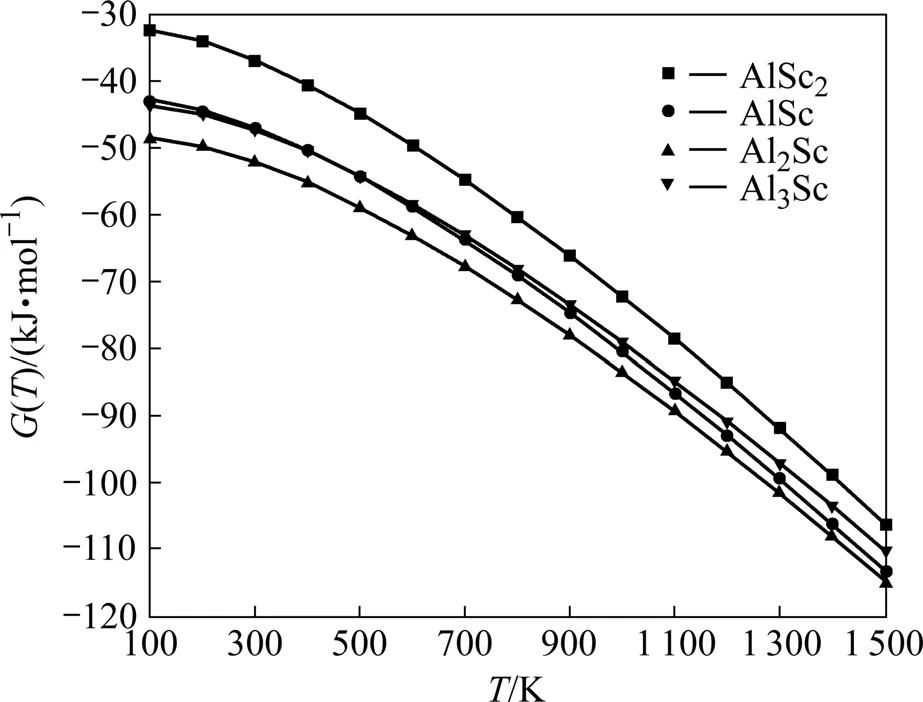
图4 Al-Sc金属间化合物的自由能—温度曲线图Fig.4 Free energy—temperature curves of Al-Sc intermetallics compounds
4 结论
1) 在密度泛函理论的广义梯度近似下,优化Al-Sc合金的 4个稳定相的晶体结构,各相成键在费米面以下以Sc 3d-Al 3s、Sc 3d-Al 3p杂化为主,在费米面以上则主要是Sc−Sc成键;Al2Sc和Al3Sc在低能级区成键较多,且能带展宽程度大,体系共价性较强,而AlSc和AlSc2在低能级区成键较少,电子局域程度较大。
2) 计算得到的体弹模量、德拜温度、生成焓和结合能等与已有试验数据吻合较好。0 K时,Al2Sc合金化的形成能力最强,AlSc2的最差;随着温度的升高,4种晶体的稳定性都降低,其中Al2Sc结构稳定性最强,AlSc2的结构稳定性最差。AlSc相比Al3Sc在低温时(400 K以下)理论上更易生成,但由于Sc的低温溶解度几乎为零,且Al3Sc的成键比AlSc的稳定,因此,实际中往往是Al3Sc析出相多于AlSc相。
REFERENCES
[1] IMAMURA T. Current status and trend of applicable material technology for aerospace structure[J]. Journal of Japan Institute of Light Metals, 1999, 49(7): 302−305.
[2] 杨少华, 邱竹贤, 张明杰. 铝钪合金的应用及生产[J]. 轻金属,2006(4): 55−57.YANG Shao-hua, QIU Zhu-xian, ZHANG Ming-jie. Application and production of Al-Sc alloy[J]. Light Metals, 2006(4): 55−57.
[3] 肖代红, 巢 宏, 陈康华, 黄伯云. 微量Sc对AA7085铝合金组织与性能的影响[J]. 中国有色金属学报, 2008, 18(12):2145−2150.XIAO Dai-hong, CHAO Hong, CHEN Kang-hua, HUANG Bai-yun. Effect of minor Sc addition on microstructure and properties of AA7085 alloy[J]. The Chinese Journal of Nonferrous Metals, 2008, 18(12): 2145−2150.
[5] KRESSE G, JOUBERT J. From ultrasoft pseudopotentials to the projector augmented-wave method[J]. Phys Rev B, 1999, 59:1758−1775.
[6] TAO Xiao-ma, OUYANG Yi-fang. Ab initio calculation of the total energy and elastic properties of Laves phase C15 Al2RE(RE = Sc, Y, La, Ce–Lu)[J]. Computational Materials Science,2008, 44(2): 392−399.
[7] TAO Xiao-ma, OUYANG Yi-fang, LIU Hua-shan, ZENG Fan-jing, FENG Yuan-ping, JIN Zhan-peng. Calculation of the thermodynamic properties of B2 AlRE (RE Sc, Y, La, Ce-Lu)[J].Physica B, 2007, 399: 27−32.
[8] LU Xiao-gang, WANG Yi. Calculation of the vibrational contribution to the Gibbs energy of formation for AlSc[J].Calphad, 2002, 26(4): 555−561.
[9] WANG Yi, CHEN Dong-quan, ZHANG Xin-wei. Calculated equation of state of Al, Cu, Ta, Mo, and W to 1 000 GPa[J]. Phys Rev Lett, 2000, 15(84): 3220−3223.
[10] ASTA M, OZOLINS V. Structural, vibrational and thermodynamic properties of Al-Sc alloys and intermetallic compounds[J]. Phys Rev B, 2001, 64: 094104−14.
[11] ASTA M, FOILES S M, QUONG A A. First-principles calculations of bulk and interfacial thermodynamic properties for fcc-based Al-Sc alloys[J]. Phys Rev B, 1998, 57:11265−11275.
[12] CACCIAMANI G, RIANI P, BORZONE G, PARODI N,SACCONE A, FERRO R, PISCH A, SCHMID-FETZER R.Thermodynamic measurements and assessment of the Al-Sc system[J]. Intermetallics, 1999, 7: 101−108.
[13] JOHN P P, WANG Yue. Accurate and simple analytic representation of the electron-gas correlation energy[J]. Phys Rev B, 1992, 45: 13244−13249.
[14] EYMOND S, PARTHÈ E, LESS-COMMON J. The Al-rich region in the Y-Ni-Al system: Microstructures and phase equilibria[J]. Met, 1969, 19: 441−448.
[15] SCHUSTER J C, BAUER J, LESS-COMMON J. Phase equilibria in M-X-X' and M-Al-X ternary systems (M=transition metal; X, X' = B, C, N, Si) and the crystal chemistry of ternary compounds[J]. Met, 1985, 109: 345−350.
[16] TAO Xiao-ma, OUYANG Yi-fang, JIN Zhan-peng. Ab initio calculations of mechanical and thermodynamic properties for the B2-based AlRE[J]. Computational Materials Science, 2007,40(2): 226−233.
[17] NGUYEN-MANH D, PETTIFOR D G. Electronic structure,phase stability and elastic moduli of AB transition metal aluminides[J]. Intermetallics, 1999, 7: 1095−1106.
[18] CHEN Xing-qiu, WOLF W, PODLOUCKY R, ROGL P.Miedema’s model revisited: The parameterψ*for Ti, Zr and Hf[J]. Intermetallics, 2004, 12: 59−64.
[19] HARADA Y. Microstructure of Al3Sc with ternary rare-earth additions[J]. Intermetallics, 2009, 17: 17−24.
[20] NYLÉN J, GARCÌA F J, MOSEL B D, PǑTTGEN R,HĀUSSERMANN U. Structure relationships, phase stability and bonding of compounds PdSnn(n=2, 3, 4)[J]. Solid State Sci,2004, 6(1): 147−155.
[21] 徐万东, 张瑞林, 余瑞靖. 过渡金属化合物晶体结合能计算[J]. 中国科学A辑, 1988(3): 323−331.XU Wan-dong, ZHANG Rui-lin, YU Rui-jing. The binding energy calculation of transition metal compounds[J]. Science in China: Serie A, 1988(3): 323−331.
[22] LI Chong-he, JOO L H, WU Ping. Empirical correlation between melting temperature and cohesive energy of binary Laves phases[J]. Journal of Physics and Chemistry of Solids, 2003, 64:201−212.
[23] 徐 慧. 凝聚态物理专题[M]. 长沙: 中南大学出版社, 2009:45−46.XU Hui. Topics of condensed matter physics[M]. Changsha:Central South University Press, 2009: 45−46.
[24] MORUZZI V L, JANAK J F, SCHWARZ K. Calculated thermal properties of metals[J]. Physical Review B, 1988, 37:790−798.
[25] MAYER B, ANTON H, BOTT E, METHFESSEL M, STICHT J,HARRIS J, SCHMIDT P C. Ab-initio calculation of the elastic constants and thermal expansion coefficients of laves phases[J].Intermetallics, 2003, 11(1): 23−32.
[26] XU Ji-hua, FREEMAN A J. Phase stability and electronic structure of ScAl3and ZrAl3and of Sc-stabilized cubic ZrAl3precipitates[J]. Phys Rev B, 1990, 41: 12553−12561.
[27] HYLAND R W, STIFFLER R C. Determination of the elastic constants of polycrystalline Al3Sc[J]. Scripta Metall Mater, 1991,25(2): 473−477.
[28] 张明杰, 李金丽, 梁家骁. 盐熔电解法生产Al-Sc合金[J]. 东北大学学报: 自然科学版, 2003, 24(4): 355−360.ZHANG Ming-jie, LI Jin-li, LIANG Jia-xiao. Preparation of Al-Sc alloys by molten salt electrolysis[J]. Journal of Northeastern University: Natural Science, 2003, 24(4): 355−360.
(编辑 杨 华)
Electronic structure, stability and thermodynamic properties of Al-Sc intermetallics compounds
LI Yan-feng1,2, XU Hui1,2, ZHANG Biao1, ZHANG Li-gang2
(1. School of Physical Science and Technology, Central South University, Changsha 410083, China;2. School of Materials Science and Engineering, Central South University, Changsha 410083, China)
1004-0609(2010)05-0946-08
湖南省自然科学基金资助项目(07JJ3102);长沙市科技计划资助项目(k0902132-11)
2009-09-18;
2010-01-26
徐 慧,教授,博士;电话:0731-88879207;E-mail: xuekb3@mail.csu.edu.cn
猜你喜欢
中学生数理化(高中版.高考理化)(2021年5期)2021-07-16
中外文摘(2021年7期)2021-04-23
中华养生保健(2020年2期)2020-11-16
发明与创新·小学生(2020年10期)2020-10-19
复旦学报(医学版)(2020年3期)2020-06-18
发明与创新·小学生(2019年12期)2019-12-05
中学生数理化·高三版(2017年1期)2017-04-20
电子制作(2016年19期)2016-08-24
制冷技术(2016年4期)2016-08-21
中国中医药现代远程教育(2014年11期)2014-08-08
