
CH3 F与O(3 P)反应机理的量子化学研究
2010-01-09 03:05冯丽霞郑慧敏
太原师范学院学报(自然科学版) 2010年2期
冯丽霞 郑慧敏 左 玉
(太原师范学院化学系,山西太原 030031)
CH3F与O(3P)反应机理的量子化学研究
冯丽霞 郑慧敏 左 玉
(太原师范学院化学系,山西太原 030031)
用量子化学从头算M P2/6-311G(d,p)方法研究了CH3F与O(3P)反应的反应机理.在QCISD(T)/6-311G(d,p)水平上精确计算了各反应物种的能量.结果表明,标题反应共存在4类反应5个反应通道,分别为抽提氢反应、抽提氟反应、消氟化氢反应和消氢反应,抽提氢反应为主反应通道.
CH3F;抽提氢反应;抽提氟反应,消氟化氢反应;消氢反应;QCISD(T)/6-311G(d,p)//M P2/6-311G(d,p)
氢氟烃(HFCs)类化合物不含氯、溴原子,臭氧消耗系数(ODP)小[1],可以作为氯氟烃类化合物(CFCs)在发泡剂、制冷剂及火焰抑制剂方面的替代物[2].但 HFCs具有强烈的红外吸收能力,温室效应系数(GWP)较高[3],在大气中的积累可能会导致气候变暖.因此,研究 HFCs在大气中的燃烧机理具有重要意义.
目前已有不少学者分别从实验及理论角度对CH3F与Cl[4,5],H[6],OH[7],CH3[8,9]等原子或自由基的反应进行了研究.综合比较发现,人们对这类反应的宏观动力学性质关注较多,给出的反应微观机理信息较少.对于标题反应,Kreye等[10]曾通过理论计算方法对CH3F与O(3P)抽提氢反应通道的动力学性质进行了研究,但他们并未揭示该反应在大气中的燃烧机理,理论上CH3F与O(3P)的反应还可能存在抽提氟、消氟化氢、消氢等通道,我们应对其进行全面研究,以确认CH3F在大气中的反应机理.另外,由于目前实验技术的限制,许多不稳定物种的生成热数据仍存在准确性低,不确定度大等问题.本文通过双水平QCISD(T)/6-311G(d,p)//M P2/6-311G(d,p)计算方法获得了标题反应的势能信息,这不仅为实验方面精确测定不稳定物种的生成热数据提供一定的理论参考,且对揭示CH3F的燃烧特性、燃烧机理,建立CH3F的高温燃烧模型、开发新型火焰抑制剂有重要意义.
1 计算方法
在M P2/6-311G(d,p)水平上全优化了抽提氢、抽提氟、消氟化氢和消氢反应通道所有驻点的几何构型,通过振动频率分析对各稳定点与过渡态的结构进行了确认,并用内禀反应坐标(IRC)方法计算确证了各过渡态与相应反应物和产物的相关性.此外,本文选用双水平QCISD(T)/6-311G(d,p)//M P2/6-311G(d,p)方法精修了所有驻点的单点能.以上全部工作均采用 Gaussian03[11]程序包完成.
2 结果与讨论
通过对CH3F与O(3P)反应机理的研究,我们得到了4类,共5个反应通道,分别为:其中,R1为抽提氢反应通道,相应过渡态 TS1,产物 P1;R2为抽提氟反应,过渡态为 TS2,产物 P2;R3为消氟化氢反应,过渡态TS3,产物P3;R4a,R4b均为消氢反应通道,对于同样的产物P4对应的过渡态分别记为TS4a,TS4b.


2.1 驻点构型与频率
图1给出了M P2/6-311G(d,p)水平上优化得到的所有反应物、产物和过渡态的几何构型及个别物种的实验值[6,12~14].对比图1数据,对反应物CH3F和产物OH,CH3,HF,理论计算的键长、键角与实验值非常接近,如:键长最大偏差仅为0.000 9 nm(C-F键),采用M P2/6-311G(d,p)方法计算各稳定点结构参数合理.反应通道上各驻点的计算频率及个别物种的实验频率[6,12,13,15,16]列于表1.表1中,M P2/6-311G(d,p)水平上计算的频率虽多数高于实验值,但最大偏差仍在6%以内,本文计算结果可靠.图2为各过渡态的虚频振动模式.
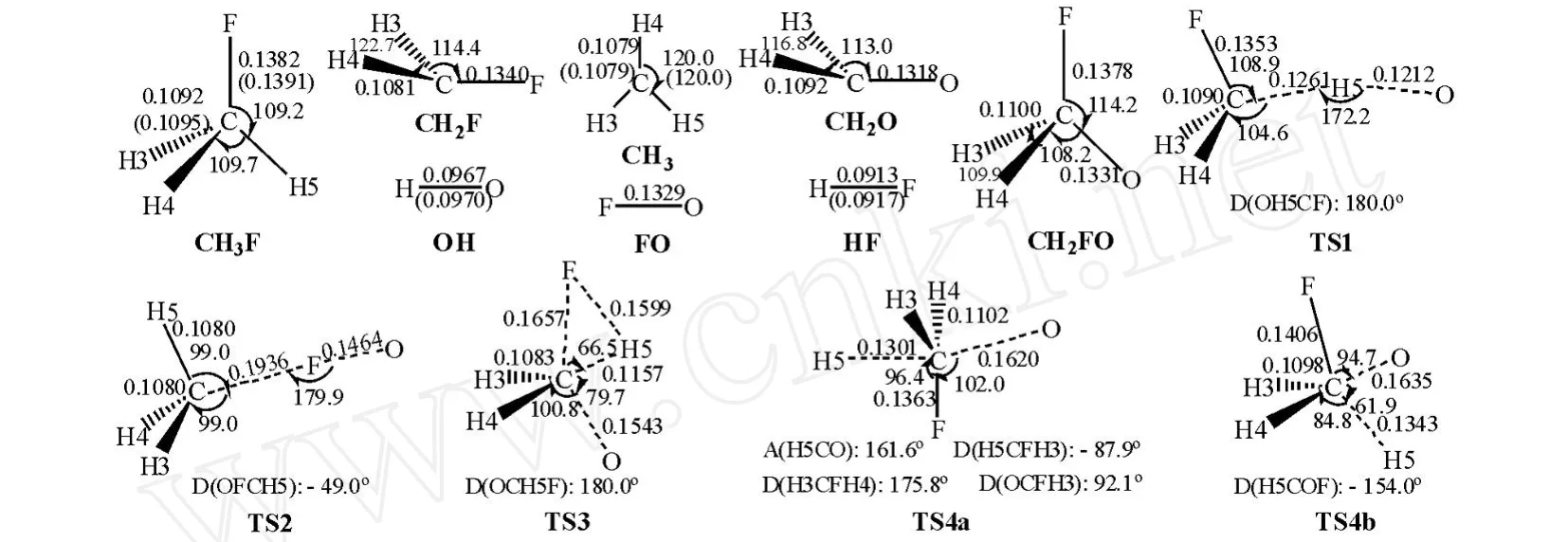
图1 M P2/6-311G(d,p)水平上优化的各反应驻点几何构型(键长:nm;键角:°)Fig.1 The optimized parameters of all the stationary points involved in the title reaction at the M P2/6-311G(d,p)level(distances in nanometers and angles in degrees)
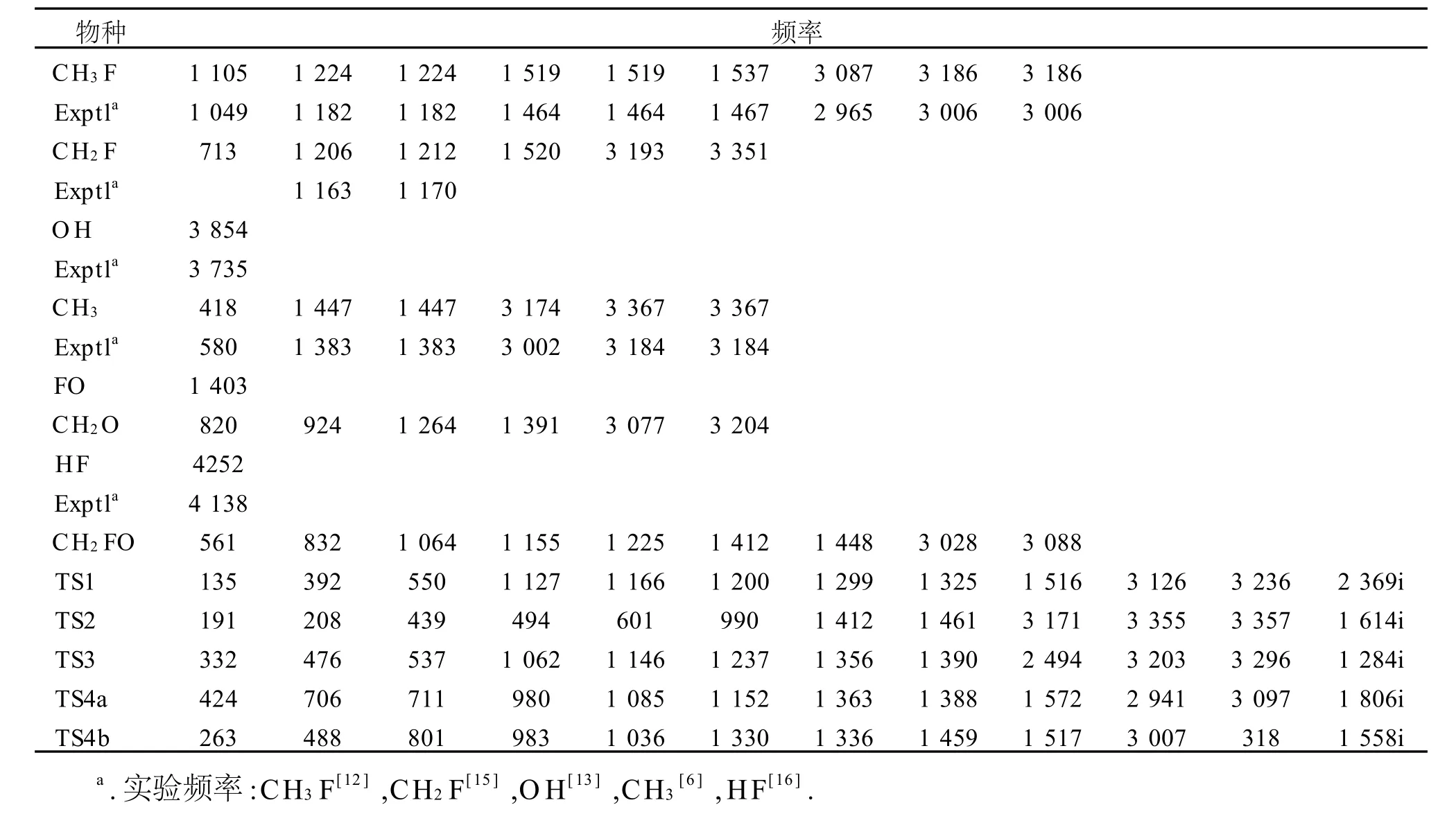
表1 M P2/6-311G(d,p)水平上计算的标题反应各驻点的谐振频率Table 1 The harmonic vibrational frequencies for the reactants,p roducts and saddle points involved in the reaction of CH3 F with O(3 P)at the MP2/6-311G(d,p)level cm-1
2.1.1 抽提氢反应
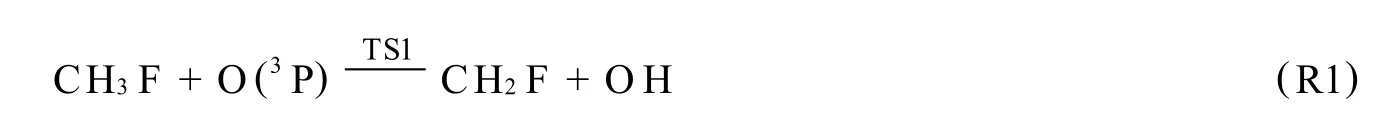
与反应物CH3F相比,过渡态 TS1的C-F键长减小了0.002 9 nm,欲断裂的C-H5键长比平衡的C-H 5键长增加了15.5%,欲形成的O-H键长是正常O-H键长的1.253倍,其余键长、键角几乎保持不变.TS1中C-F键的减少或许是由于活化键C-H5增长后导致 FCH5间空间位阻降低所致.由频率计算知,TS1有唯一虚频2369i,其振动的合运动效果表现为 H5在C与O原子间的运动,此振动模式与相应过渡态走向反应物或产物的位移向量相对应,说明 TS1确是抽提氢反应通道的合理过渡态.
2.1.2 抽提氟反应
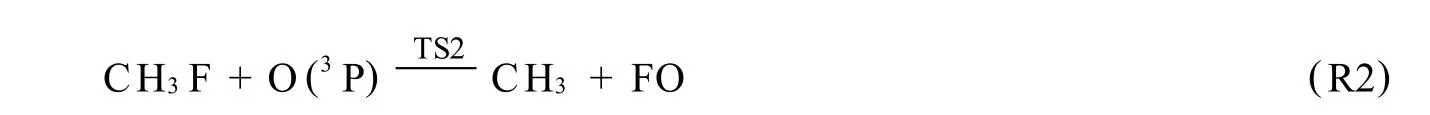
与反应物CH3F相比,过渡态TS2中欲断裂的C-F键长比平衡的C-F键长增加了40.0%,欲形成的FO键长是产物FO键长的1.102倍.此外,键角 FCH5比CH3F中相应键角减少了10.2°.TS2有唯一虚频1614i,其振动模式主要为F在C,O原子间的运动.
2.1.3 消氟化氢反应

过渡态 TS3中C-F,C-H 5,C-O,H-F的键长分别是反应物CH3F,产物CH2O,HF中相应键长的1.199,1.060,1.171,1.751倍;TS3有唯一虚频1 284i cm-1,其虚频振动模式主要是C原子在两个振动幅度较小的F,O原子间的运动,同时体系伴随着 H 5原子向F原子靠近时,C原子向O原子靠近;H5远离F时,C也开始远离O的运动,其中 H原子的运动幅度远较F,O两原子的大.
2.1.4 消氢反应

在消氢反应中,本文共找到两个过渡态 TS4a,TS4b.对比发现,通道 R4a是典型的SN2型取代反应.过渡态 TS4a中,O原子与C,H5原子近似处在同一直线上(键角 H5CO为161.6°),同时被O原子进攻的CH2F基团中的四个原子C,H 3,H 4与 F则几乎处在垂直于这条线的同一平面上(二面角 H 5CFH3为-87.9°,OCFH3为92.1°,二面角 H3CFH4为175.8°),此结构中欲断裂的C-H5键长比反应物中相应键长增加了19.1%;欲形成的C-O键长是相应产物中C-O键的1.217倍.TS4a有唯一虚频1806i cm-1,其振动的合运动效果表现为C在H5,O原子间的运动,同时伴随CH2F基团的往复翻转,即随着O向C靠近,C-F,C-H 3,C-H 4键向 H5原子端翻转,同时将远端的 H5原子逐出.在 TS4b中,欲断裂的C-H 5键长比反应物中相应键长增加了23.0%,欲形成的F-O键长是产物CH2FO中相应键长的1.228倍.TS4b有唯一虚频1558icm-1,其虚频对应的振动模式为O向C靠近时 H5远离C;O远离C时 H5接近C.

图2 过渡态的虚频振动模式Fig.2 The imaginary vibrationalmodes of the transition states
2.2 驻点能量
表2列出了QCISD(T)/6-311G(d,p)//M P2/6-311G(d,p)水平上计算的各反应通道经M P2/6-311G(d,p)水平零点能校正的反应能ΔE、反应能垒ΔE≠.图3是据表2的能量结果绘出的反应势能剖面图.由表2数据知,在双水平QCISD(T)/6-311G(d,p)//M P2/6-311G(d,p)上,5个反应通道的反应能ΔE分别是14.8、258.0、-66.3、20.0和20.0 kJ·mol-1,其中消氟化氢反应放热,其余三类反应抽提氢、抽提氟、消氢反应均吸热;相应反应能垒ΔE≠分别为57.0、321.4、228.5、230.4和271.7 kJ·mol-1.结合图3的势能剖面图,可进一步看出抽提氢反应的能垒比其余3类反应低得多,故其为标题反应的主反应通道.另外,对比同样的消去氟原子的反应R2(抽提氟)、R3(消氟化氢)的能量知,消氟化氢反应通道R3中不仅过渡态的能量(ΔE≠为228.5 kJ·mol-1)远较 R2(321.4 kJ·mol-1)的低 ,且生成的产物 P3(ΔE为 -66.3 kJ·mol-1)也远比产物P2稳定(258.0 kJ·mol-1).事实上,从 R2中很高的ΔE≠、ΔE数据也可看出,通道R2的过渡态很难形成,即使R2反应能克服321.4 kJ·mol-1的能垒,生成的产物 P2(258.0 kJ·mol-1)也极不稳定,故我们可认为即使在高温大气中,R2反应发生的几率也不会太高,标题反应要想消去氟原子只能发生消氟化氢反应.此外,比较产物的能量信息知,产物P3是四类反应中生成的最稳定产物.

表2 经M P2/6-311G(d,p)零点能校正的反应能ΔE、反应能垒ΔE≠Table 2 The reaction energiesΔE and the potential barriersΔE≠with the M P2/6-311G(d,p)ZPE correction at the QCISD(T)/6-311 G(d,p)level kJ·mol-1

图3 CH3 F与O(3 P)反应势能剖面示意图Fig.3 Energetic p rofile of the po tential energy surface
2.3 反应路径特性
图4列出了M P2(full)/6-311G(d,p)水平上,步长为0.05(amu)1/2·bohr时各类反应键长沿反应坐标s变化图.为便于比较,图4-c′还列出了消氟化氢反应中部分键角随s变化图.
抽提氢反应键长沿反应坐标s的变化如图4-a所示,从s=-1.0(amu)1/2(bohr开始,欲断裂的键CH 5呈线性急剧增长,到s=0.7(am u)1/2·bohr时,已增加到0.150 nm,说明C-H 5已经接近断裂.另一方面,随着反应进行,欲形成O-H键的两个原子迅速靠近,当s= -1.0(am u)1/2·bohr时,O-H距离为0.170 nm,处于弱相互吸引状态,到s=0.7(amu)1/2·bohr时,O-H距离减小到0.100 nm左右,已接近正常键长0.097 0 nm[13].其余键长在反应过程中变化很小.抽提氟反应(图4-b)、消氢反应(图4-da、4-db)键长沿反应坐标s的变化情况与抽提氢反应类似:即整个反应过程中,除活化键发生明显变化外,其余键长几乎保持不变.
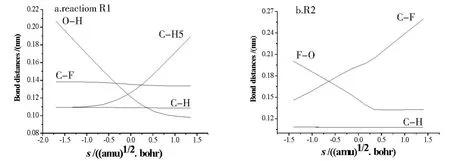
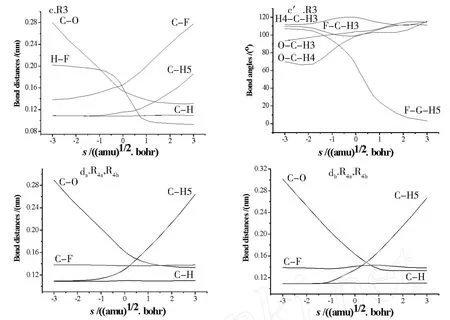
图4 M P2/6-311G(d,p)水平上计算的键长及部分键角随反应坐标s(amu)1/2·bohr变化图Fig.4 The variation of bond distances and certain bond angles as functions of s(amu)1/2·bohr at the M P2/6-311G(d,p)level
消氟化氢反应键长、键角沿反应坐标s的变化如图4-c、4-c′所示.随着反应进行,欲形成C-O键的两个原子迅速靠近,当s=1.7(am u)1/2·bohr时,C-O距离减小到0.134 nm左右,已接近产物中相应键长0.131 8 nm;而欲脱去的两个原子 H5,F与C原子之间的键长变化在起始阶段并不同步,C-F键长在反应坐标s=-2.0(amu)1/2·bohr时开始逐渐增大,而C-H5键长在s=-0.9(amu)1/2·bohr时才开始缓慢增长,经过过渡态后,C-F与C-H5开始迅速平行线性增长.此外,H5与F原子间的距离在反应初始阶段变化很小,经过过渡态后两原子间距才开始近似线性减小,当s到达1.7(amu)1/2·bohr时,H7-F键已稳定在0.095 nm左右,接近 HF分子的键长0.0917 nm[14].结合键角变化趋势(图4-c′)知,体系自s= -2.0(amu)1/2·bohr C-F键缓慢增大开始,键角FCH5即在逐渐减小,当经过过渡态C-F,C-H5迅速平行线性增长时,键角FCH5才迅速递减.整个过程可归纳为:体系自C-F键缓慢增大开始,即在调整键角 FCH5使其逐渐减小,以利于欲脱去的 H5,F原子相互靠近,产生吸引作用,到过渡态后,C-F,C-H5键开始显著拉长,键角FCH 5迅速降低,H5,F原子快速靠近,最终形成 HF分子.
3 结论
本文通过CH3F与O(3P)微观反应机理的研究,得出以下结论:
1)本文共找到4类反应的5个反应通道,分别为抽提氢反应(R1)、抽提氟反应(R2)、消氟化氢反应(R3)和消氢反应(R4a和R4b).
2)在双水平QCISD(T)/6-311G(d,p)//M P2/6-311G(d,p)上,5个反应通道的反应能ΔE分别是14.8,258.0,-66.3,20.0 和 20.0 kJ·mol-1;相应反应能垒ΔE≠分别为 57.0、321.4、228.5、230.4 和271.7 kJ·mol-1,抽提氢反应为主反应通道,消氟化氢反应通道的产物P3最稳定.
[1] Farman JD,Gardiner B G,Shanklin JD.Large lossesof total ozone in Antarctica reveal seasonal ClOx/NOx interaction[J].Nature,1985,315:207-210
[2] Houghton J T,Meria Filho L G,Callander B A,et al.Climmate change 1995:the science of climate change[M].Cambridge:Cambridge University Press,1996
[3] Finlayson-Pitts B J,Pitts J N Jr.Chemistry of the upper and low er atmosphere:theo ry,experiments,and app lications[M].San Diego:Academic Press,1999
[4] Hitsuda K,Takahashi K,Matsum i Y.Kinetics of the Reactions of Cl(2P1/2)and Cl(2P3/2)A tom s with C2H6,C2D6,CH3F,C2H5F,and CH3CF3at 298 K[J].J Phys Chem A,2001,105:5 131-5 136
[5] Rosenman E,M c Kee M L.Reaction-path dynamics and theoretical rate constants for the CH3F+Cl→HCl+CH2F Reaction by direct dynamicsmethod[J].J Am Chem Soc,1997,119:9 033-9 038
[6] Maity D K,Duncan W T,Truong T N.Direct abinitio dynamics studies of the hydrogen abstraction reactions of hydrogen atom with fluo romethanes[J].J Phys Chem A,103:2 152-2 159
[7] Lien P Y,You R M,Hu W P.Theo retical modeling of the hydrogen abstraction reaction of fluoromethane by the hydroxyl radical[J].J Phys Chem A,2001,105:2 391-2 400
[8] Raal FA,Steacie EW R.The reaction of methyl radicals with some halogenated methanes[J].JChem Phys,1952,20(4):578-581
[9] 冯丽霞,王文亮,李 琳,等.CH4-nFn(n=1~3)与CH3氢抽提反应微观动力学的理论研究 [J].高等学校化学学报,2006,27(9):1 733-1 737
[10] Kreye W C,Seybold P G.A binitio study of the energetics and thermodynamicsof hyd rogen abstraction from fluoromethanes by O(3P).Ⅱ:CFnH4-n+O(3P) →CFnH4-n…O →.CFnH4-n…O+.OH(n=0,1,2)[J].Chemical Physics Letters,2001,335:257-264
[11] Frisch M J,Trucks GW,Schlegel H B,et al.Gaussian 03,Revision A.01[Z].Pittsburgh PA:Gaussian Inc,2003
[12] Stull D R,Prohet H.JANAF thermochemical tables 2nd ed.[M].National Standard Reference Data Series N0 37,National Bureau of Standards,US Government,Washington DC:Printing Office,1971
[13] Zhang Q Z,Zhang R Q,Gu Y S.Kinetics and mechanism of O(3P)reaction with CH3CHF2:a theo retical study[J].J.Phys Chem A,2004,108:1 064-1 068
[14] Huber K P,Herzberg G.Molecular spectra andmolecular structure IV,constantsof diatomicmolecules[M].New York:Van Nostrand Reinhold Co,1979
[15] Li Q S,Yang J,Zhang SW.Reaction-path dynamicsand theoretical rate constants for the CHnF4-n+O3→HOOO+CHn-1F4-n(n=2,3)Reactions[J].J Phys Chem A,2006,110:11 113-11 119
[16] Le Roy R J.Imp roved parameterization from combined isotopomer analysisof diatomic spectra and Itsapplication to HF and DF[J].J Mol Spect,1999,194:189-196
Quantum Chem istry Study on the Reaction Mechan ism of CH3F with O(3P)
Feng Lixia Zheng Huimin Zuo Yu
(Department of Chemistry,Taiyuan Normal University,Taiyuan 030031,China)
Ab initio calculation M P2/6-311G(d,p)is used to study the reaction mechanism of CH3F with O(3P).The single-point energiesare further calculated at the QCISD(T)/6-311G(d,p)level.It is shown that the title reaction owns four typesof reactions including five reaction channels.They are the hydrogen abstraction reaction,the fluorine abstraction reaction,the HF elimination reaction and the hydrogen elimination reaction.The hydrogen abstraction reaction is the dominant channel.
CH3F;hydrogen abstraction reaction;fluorine abstraction reaction;HF elimination reaction;hydrogen elimination reaction;QCISD(T)/6-311G(d,p)//M P2/6-311G(d,p)
【责任编辑:王映苗】
1672-2027(2010)02-0090-06
O641
A
2010-01-19
冯丽霞(1981-),女,硕士,山西定襄人,太原师范学院化学系讲师,主要从事量子化学理论研究.
猜你喜欢
化工管理(2022年13期)2022-12-02
高中数理化(2022年16期)2022-09-14
北京航空航天大学学报(2022年5期)2022-06-06
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
浙江化工(2019年9期)2019-01-21
电脑知识与技术(2018年3期)2018-03-21
中国特种设备安全(2018年12期)2018-03-15
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
